

Novel pathogenic variant combination in LPL causing familial chylomicronemia syndrome in an Asian family and experimental validation in vitro: a case report
- PMID: 36345447
- PMCID: PMC9636460
- DOI: 10.21037/tp-22-15
Novel pathogenic variant combination in LPL causing familial chylomicronemia syndrome in an Asian family and experimental validation in vitro: a case report
Abstract
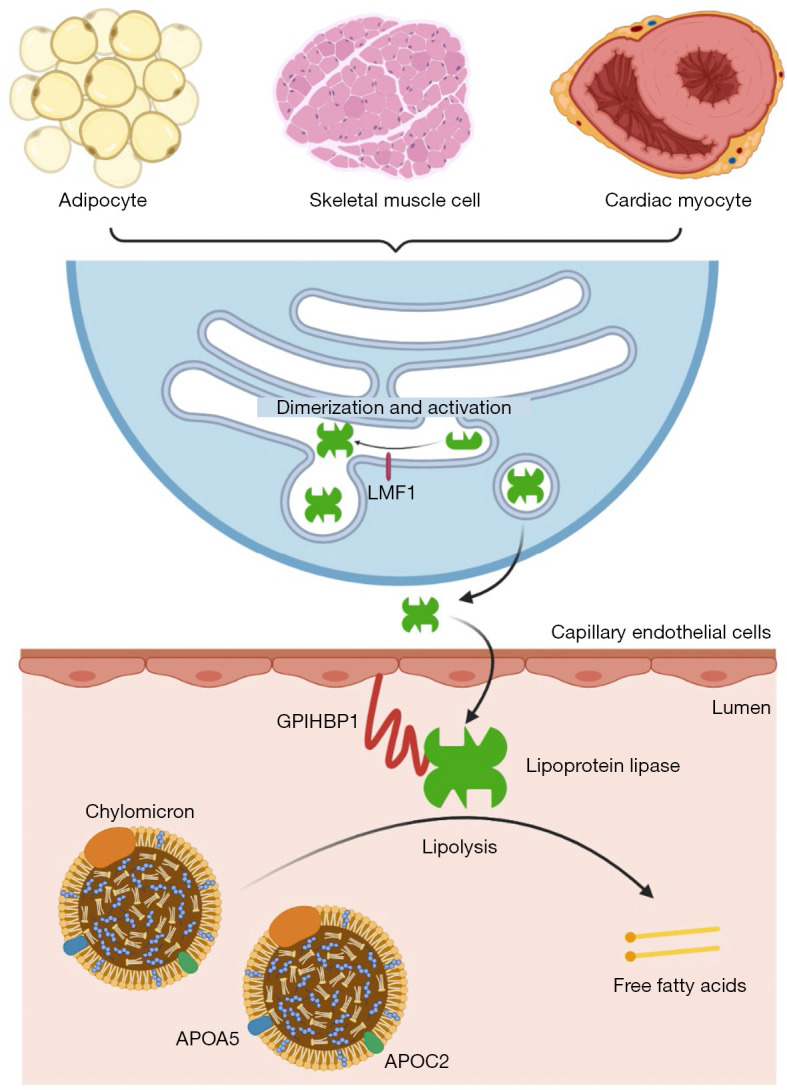
Background: Familial chylomicronemia syndrome (FCS) is a rare autosomal recessive disorder, typically caused by biallelic pathogenic variants in the lipoprotein lipase (LPL) gene. Lipoprotein lipase, encoded by the LPL gene, catalyzes the hydrolysis of triglycerides, and its deficiency or dysfunction can lead to chylomicronemia and potentially fatal recurrent acute pancreatitis.
Case description: Here, we report an Asian child with FCS due to compound heterozygous LPL variants. The 4-year-old patient presented with splenomegaly and severe hypertriglyceridemia, specifically chylomicronemia which resulted in abnormal coagulation measured by a turbidity-based assay. Based on the clinical features and family history, the diagnosis of FCS was suspected, and confirmed by the identification of compound heterozygous variants in the LPL gene (c.461A>G; p.His154Arg and c.788T>A; p.Leu263Gln) in the patient, inheriting one from each parent. According to the clinical and genetic findings, the patient was diagnosed with FCS. In vitro experimental validation found that the LPL p.H154R variant reduced the expression of lipoprotein lipase and decreased its lipolytic activity, while the LPL p.L263Q variant mainly impaired its lipolytic activity.
Conclusions: FCS was molecularly diagnosed using whole exome sequencing in the case presented. When interpreting abnormal coagulation profiles measured by turbidity-based assay, the possibility of lipemic blood (or chylomicronemia) should be considered and the presence of this phenomenon might indicate the diagnosis of FCS. In vitro experiments showed that the two LPL variants impaired lipoprotein lipase expression and/or function making them likely to be pathogenic.
Keywords: LPL gene; case report; familial chylomicronemia syndrome; lipoprotein lipase; novel pathogenic variant.
2022 Translational Pediatrics. All rights reserved.
Conflict of interest statement
Conflicts of Interest: Both authors have completed the ICMJE uniform disclosure form (available at https://tp.amegroups.com/article/view/10.21037/tp-22-15/coif). The authors report that this study was funded by Jiangsu Provincial Special Program of Medical Science (BL2012005); Jiangsu Province’s Key Medical Center (ZX201102); The Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The authors have no other conflicts of interest to declare.
Figures


Comment in
-
Familial chylomicronemia syndrome in children: a diagnosis challenge.Transl Pediatr. 2022 Nov;11(11):1743-1747. doi: 10.21037/tp-22-515. Transl Pediatr. 2022. PMID: 36506766 Free PMC article. No abstract available.
References
Publication types
LinkOut - more resources
Full Text Sources