

KLF13 Loss-of-Function Mutations Underlying Familial Dilated Cardiomyopathy
- PMID: 36346048
- PMCID: PMC9750085
- DOI: 10.1161/JAHA.122.027578
KLF13 Loss-of-Function Mutations Underlying Familial Dilated Cardiomyopathy
Abstract
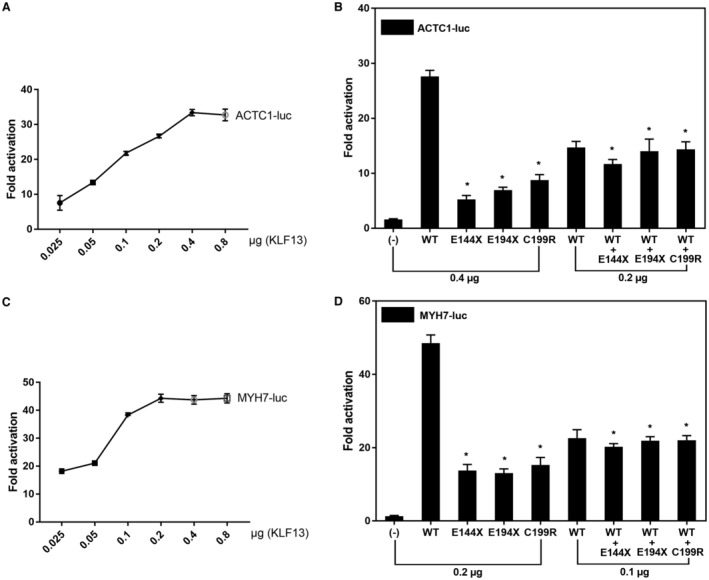
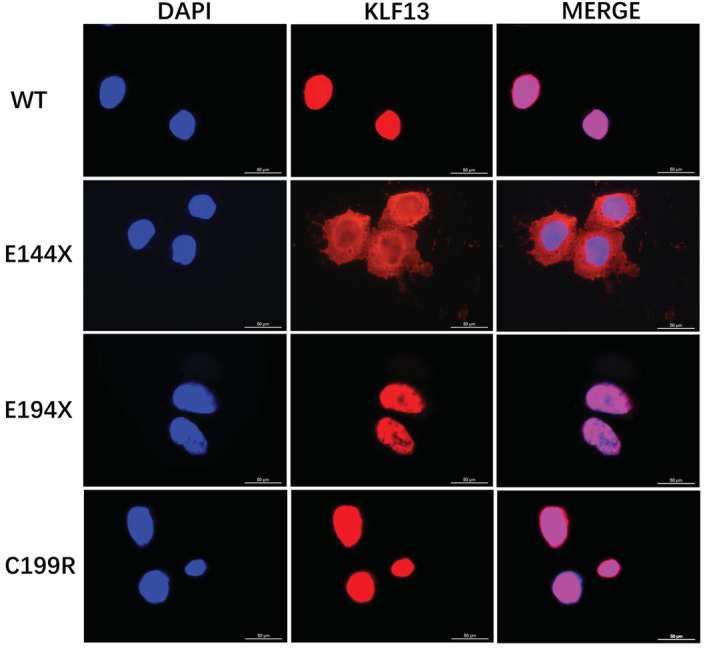
Background Dilated cardiomyopathy (DCM), characterized by progressive left ventricular enlargement and systolic dysfunction, is the most common type of cardiomyopathy and a leading cause of heart failure and cardiac death. Accumulating evidence underscores the critical role of genetic defects in the pathogenesis of DCM, and >250 genes have been implicated in DCM to date. However, DCM is of substantial genetic heterogeneity, and the genetic basis underpinning DCM remains elusive in most cases. Methods and Results By genome-wide scan with microsatellite markers and genetic linkage analysis in a 4-generation family inflicted with autosomal-dominant DCM, a new locus for DCM was mapped on chromosome 15q13.1-q13.3, a 4.77-cM (≈3.43 Mbp) interval between markers D15S1019 and D15S1010, with the largest 2-point logarithm of odds score of 5.1175 for the marker D15S165 at recombination fraction (θ)=0.00. Whole-exome sequencing analyses revealed that within the mapping chromosomal region, only the mutation in the KLF13 gene, c.430G>T (p.E144X), cosegregated with DCM in the family. In addition, sequencing analyses of KLF13 in another cohort of 266 unrelated patients with DCM and their available family members unveiled 2 new mutations, c.580G>T (p.E194X) and c.595T>C (p.C199R), which cosegregated with DCM in 2 families, respectively. The 3 mutations were absent from 418 healthy subjects. Functional assays demonstrated that the 3 mutants had no transactivation on the target genes ACTC1 and MYH7 (2 genes causally linked to DCM), alone or together with GATA4 (another gene contributing to DCM), and a diminished ability to bind the promoters of ACTC1 and MYH7. Add, the E144X-mutant KLF13 showed a defect in intracellular distribution. Conclusions This investigation indicates KLF13 as a new gene predisposing to DCM, which adds novel insight to the molecular pathogenesis underlying DCM, implying potential implications for prenatal prevention and precision treatment of DCM in a subset of patients.
Keywords: KLF13; biological assay; dilated cardiomyopathy; functional genomics; molecular genetics; transcriptional factor.
Figures





Similar articles
-
KLF13 loss-of-function variation contributes to familial congenital heart defects.Eur Rev Med Pharmacol Sci. 2020 Nov;24(21):11273-11285. doi: 10.26355/eurrev_202011_23617. Eur Rev Med Pharmacol Sci. 2020. PMID: 33215447
-
VEZF1 loss-of-function mutation underlying familial dilated cardiomyopathy.Eur J Med Genet. 2023 Mar;66(3):104705. doi: 10.1016/j.ejmg.2023.104705. Epub 2023 Jan 16. Eur J Med Genet. 2023. PMID: 36657711
-
ZBTB17 loss-of-function mutation contributes to familial dilated cardiomyopathy.Heart Vessels. 2018 Jul;33(7):722-732. doi: 10.1007/s00380-017-1110-4. Epub 2018 Feb 14. Heart Vessels. 2018. PMID: 29445930
-
[Genetics of dilated cardiomyopathy].Z Kardiol. 2001 Jul;90(7):461-9. doi: 10.1007/s003920170134. Z Kardiol. 2001. PMID: 11515275 Review. German.
-
Polymorphisms in genes encoding nonsarcomeric proteins and their role in the pathogenesis of dilated cardiomyopathy.Herz. 2012 Dec;37(8):836-41. doi: 10.1007/s00059-012-3698-6. Herz. 2012. PMID: 23188159 Review.
Cited by
-
Investigating possible dilated cardiomyopathy targets via bioinformatic analysis.Am J Transl Res. 2023 Jul 15;15(7):4788-4795. eCollection 2023. Am J Transl Res. 2023. PMID: 37560246 Free PMC article.
-
Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias.Biology (Basel). 2023 Feb 21;12(3):346. doi: 10.3390/biology12030346. Biology (Basel). 2023. PMID: 36979038 Free PMC article.
-
Drug development advances in human genetics-based targets.MedComm (2020). 2024 Feb 9;5(2):e481. doi: 10.1002/mco2.481. eCollection 2024 Feb. MedComm (2020). 2024. PMID: 38344397 Free PMC article. Review.
-
Krüppel-like Factor-9 and Krüppel-like Factor-13: Highly Related, Multi-Functional, Transcriptional Repressors and Activators of Oncogenesis.Cancers (Basel). 2023 Nov 30;15(23):5667. doi: 10.3390/cancers15235667. Cancers (Basel). 2023. PMID: 38067370 Free PMC article. Review.
-
Putting transcriptional brakes on fibrosis: a negative regulator of TGFβ signaling.Trends Cell Biol. 2023 Sep;33(9):734-735. doi: 10.1016/j.tcb.2023.06.003. Epub 2023 Jun 26. Trends Cell Biol. 2023. PMID: 37380582 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
