

Swiss public health measures associated with reduced SARS-CoV-2 transmission using genome data
- PMID: 36346321
- PMCID: PMC9765449
- DOI: 10.1126/scitranslmed.abn7979
Swiss public health measures associated with reduced SARS-CoV-2 transmission using genome data
Abstract
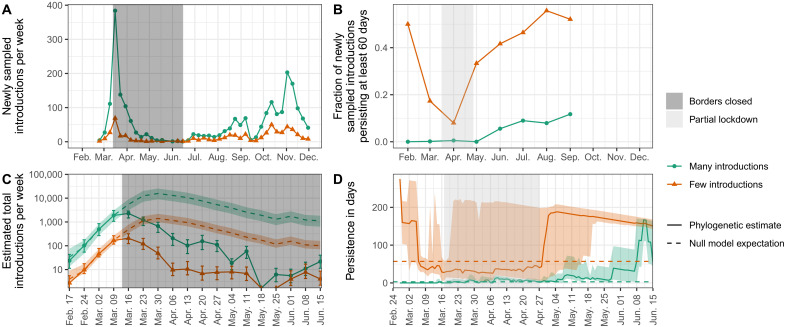
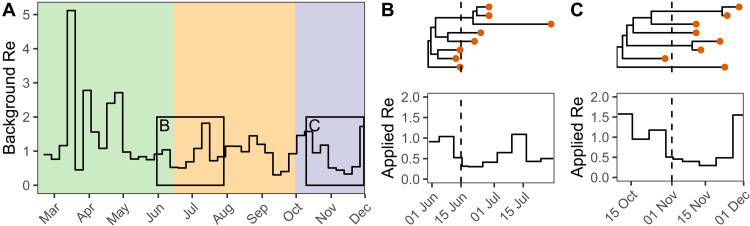
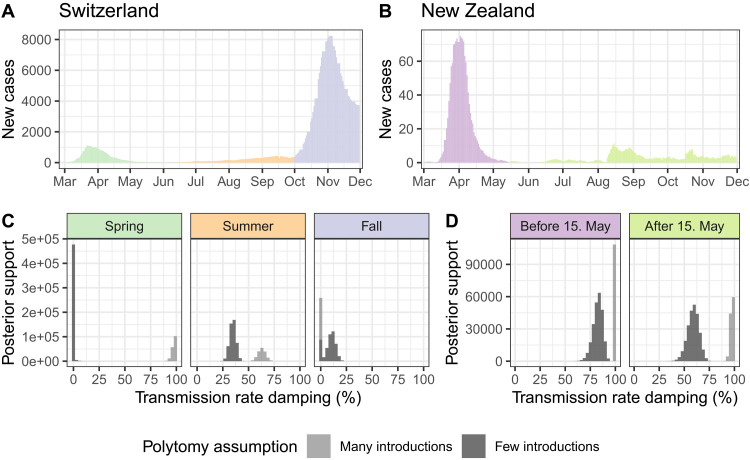
Genome sequences from evolving infectious pathogens allow quantification of case introductions and local transmission dynamics. We sequenced 11,357 severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomes from Switzerland in 2020-the sixth largest effort globally. Using a representative subset of these data, we estimated viral introductions to Switzerland and their persistence over the course of 2020. We contrasted these estimates with simple null models representing the absence of certain public health measures. We show that Switzerland's border closures decoupled case introductions from incidence in neighboring countries. Under a simple model, we estimate an 86 to 98% reduction in introductions during Switzerland's strictest border closures. Furthermore, the Swiss 2020 partial lockdown roughly halved the time for sampled introductions to die out. Last, we quantified local transmission dynamics once introductions into Switzerland occurred using a phylodynamic model. We found that transmission slowed 35 to 63% upon outbreak detection in summer 2020 but not in fall. This finding may indicate successful contact tracing over summer before overburdening in fall. The study highlights the added value of genome sequencing data for understanding transmission dynamics.
Figures
References
-
- Oude Munnink B. B., Worp N., Nieuwenhuijse D. F., Sikkema R. S., Haagmans B., Fouchier R. A. M., Koopmans M., The next phase of SARS-CoV-2 surveillance: Real-time molecular epidemiology. Nat. Med. 27, 1518–1524 (2021). - PubMed
-
- Rockett R. J., Arnott A., Lam C., Sadsad R., Timms V., Gray K.-A., Eden J.-S., Chang S., Gall M., Draper J., Sim E. M., Bachmann N. L., Carter I., Basile K., Byun R., O’Sullivan M. V., Chen S. C.-A., Maddocks S., Sorrell T. C., Dwyer D. E., Holmes E. C., Kok J., Prokopenko M., Sintchenko V., Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Nat. Med. 26, 1398–1404 (2020). - PubMed
-
- Douglas J., Mendes F. K., Bouckaert R., Xie D., Jiménez-Silva C. L., Swanepoel C., de Ligt J., Ren X., Storey M., Hadfield J., Simpson C. R., Geoghegan J. L., Drummond A. J., Welch D., Phylodynamics reveals the role of human travel and contact tracing in controlling the first wave of COVID-19 in four island nations. Virus Evol. 7, veab052 (2021). - PMC - PubMed
-
- Mallon P. W. G., Crispie F., Gonzalez G., Tinago W., Garcia Leon A. A., McCabe M., de Barra E., Yousif O., Lambert J. S., Walsh C. J., Kenny J. G., Feeney E., Carr M., Doran P., Cotter P. D., Whole-genome sequencing of SARS-CoV-2 in the Republic of Ireland during waves 1 and 2 of the pandemic. medRxiv , 2021.02.09.21251402 (2021).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
