

Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats
- PMID: 36346652
- PMCID: PMC9643004
- DOI: 10.7554/eLife.79777
Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats
Abstract
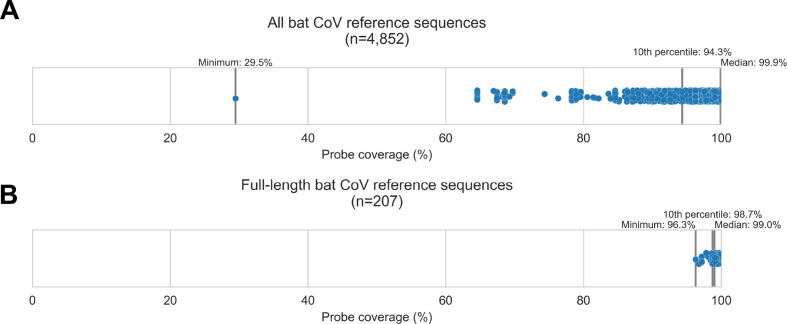
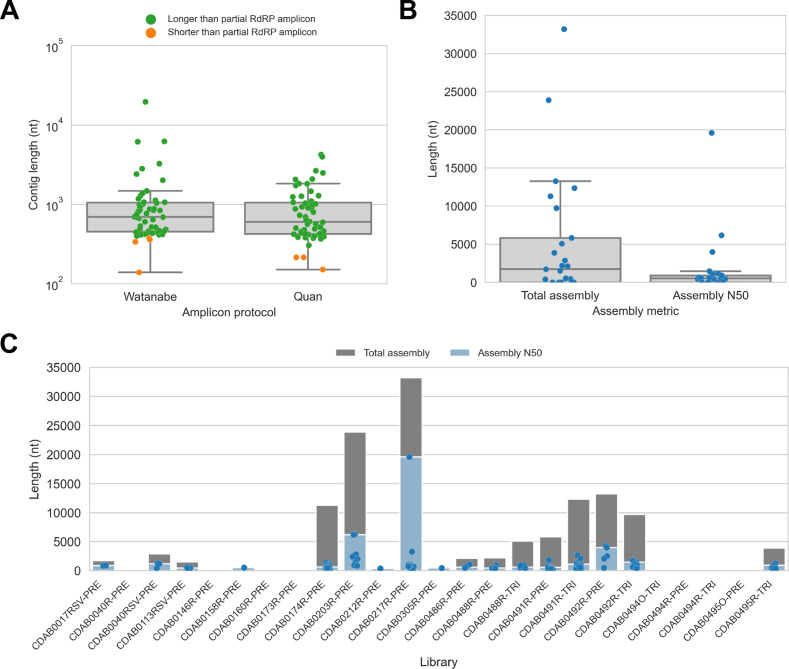
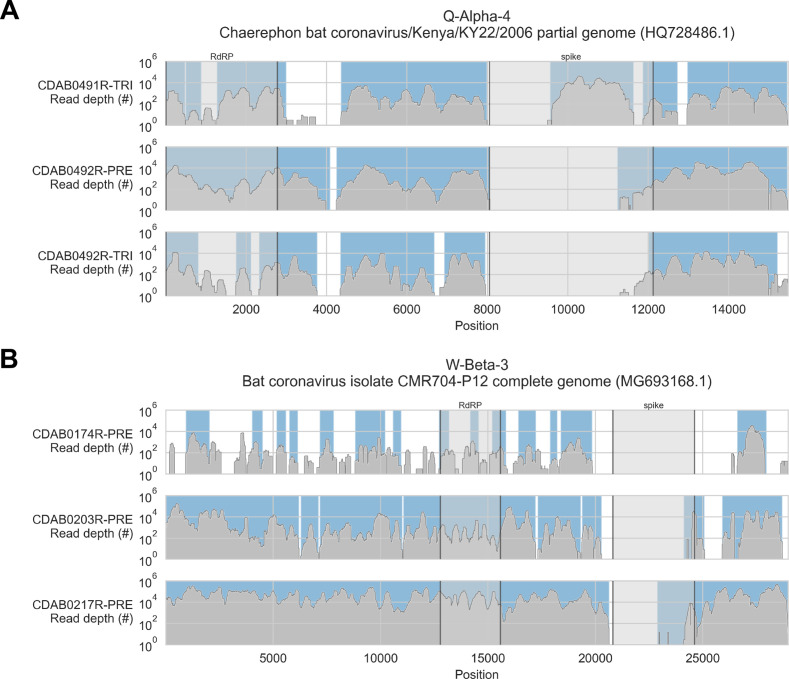
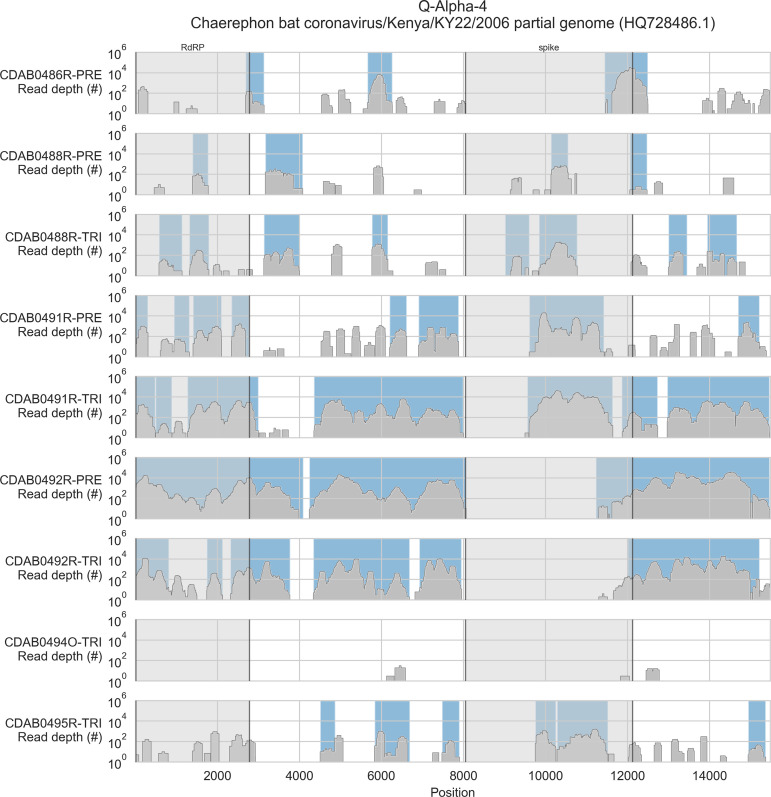
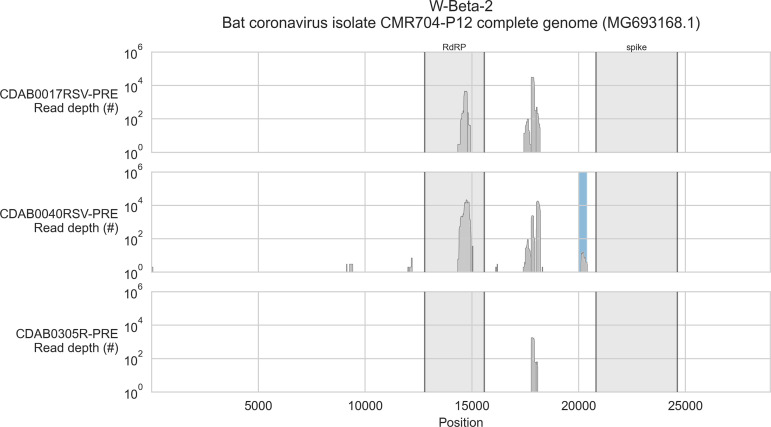
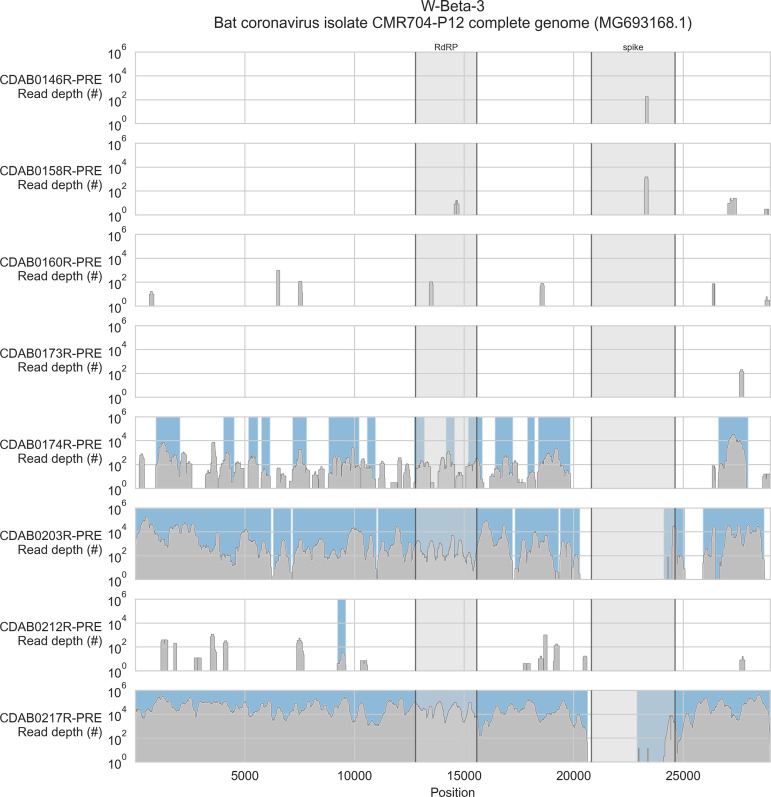
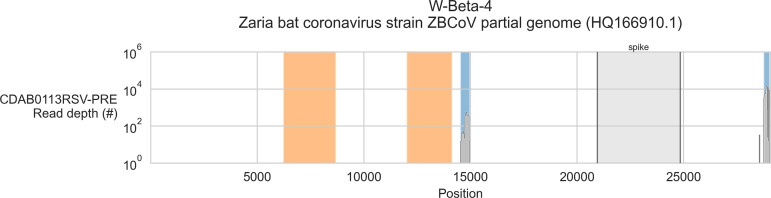
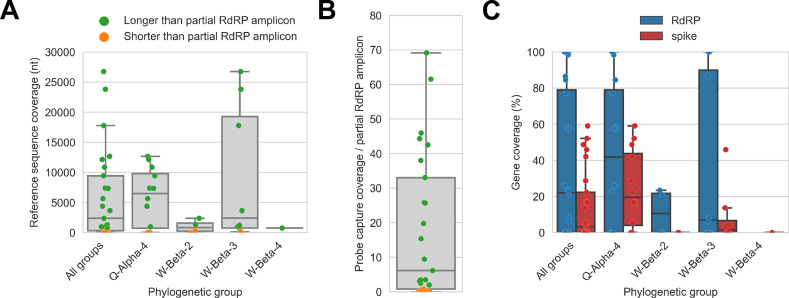
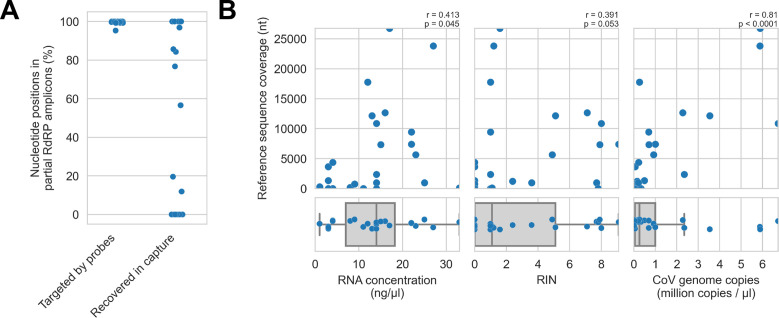
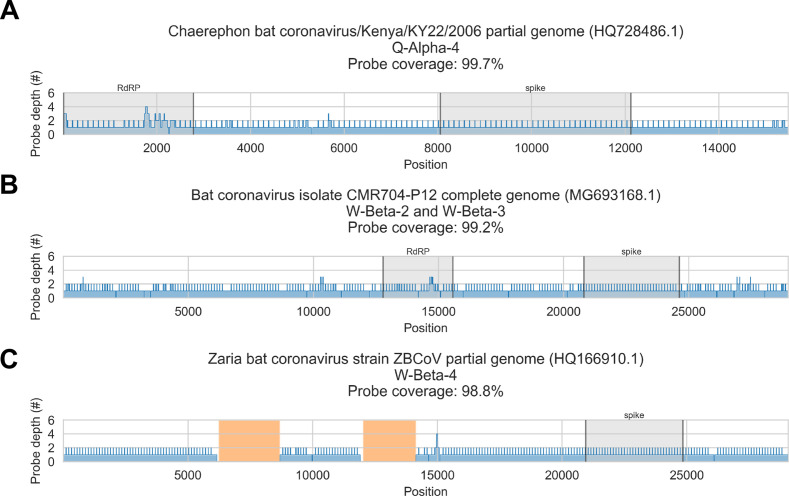
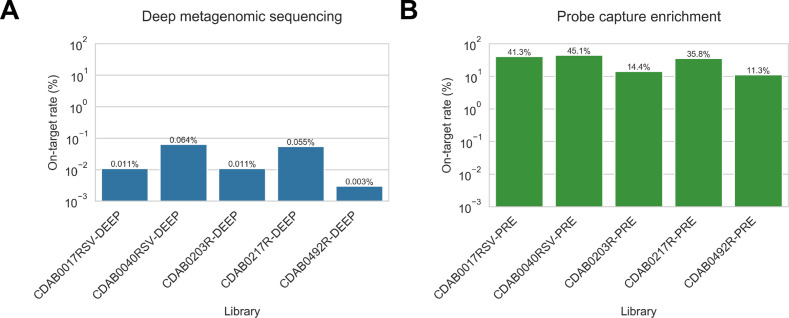
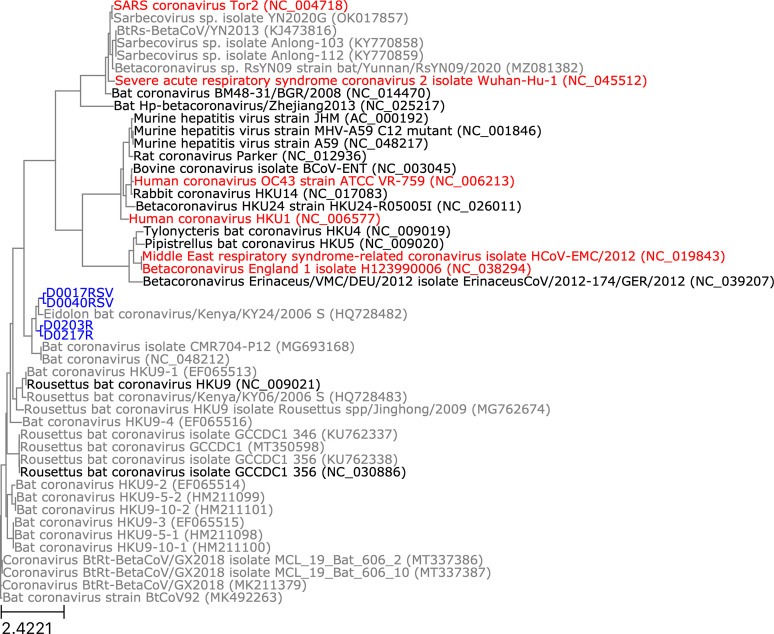
Public health emergencies like SARS, MERS, and COVID-19 have prioritized surveillance of zoonotic coronaviruses, resulting in extensive genomic characterization of coronavirus diversity in bats. Sequencing viral genomes directly from animal specimens remains a laboratory challenge, however, and most bat coronaviruses have been characterized solely by PCR amplification of small regions from the best-conserved gene. This has resulted in limited phylogenetic resolution and left viral genetic factors relevant to threat assessment undescribed. In this study, we evaluated whether a technique called hybridization probe capture can achieve more extensive genome recovery from surveillance specimens. Using a custom panel of 20,000 probes, we captured and sequenced coronavirus genomic material in 21 swab specimens collected from bats in the Democratic Republic of the Congo. For 15 of these specimens, probe capture recovered more genome sequence than had been previously generated with standard amplicon sequencing protocols, providing a median 6.1-fold improvement (ranging up to 69.1-fold). Probe capture data also identified five novel alpha- and betacoronaviruses in these specimens, and their full genomes were recovered with additional deep sequencing. Based on these experiences, we discuss how probe capture could be effectively operationalized alongside other sequencing technologies for high-throughput, genomics-based discovery and surveillance of bat coronaviruses.
Keywords: DNA sequencing; bat; coronavirus; genome; infectious disease; microbiology; probe capture; viruses.
© 2022, Kuchinski et al.
Conflict of interest statement
KK, KL, DS, JR, AS, ML, NL, JM, NP, AC No competing interests declared, CK, FM, PM, IN, FN, JA, JA, CM, ER, DM were employed by Metabiota Inc, MM, KS, CL are employees of Labyrinth Global Health Inc and were employed by Metabiota Inc, AG is an employee of Development Alternatives Inc and was employed by Metabiota Inc, DJ is an employee of Nyati Health Consulting and was employed by Metabiota Inc, NW is an employee of Metabiota Inc
Figures

References
-
- Bonsall D, Ansari MA, Ip C, Trebes A, Brown A, Klenerman P, Buck D, Piazza P, Barnes E, Bowden R, STOP-HCV Consortium Ve-SEQ: robust, unbiased enrichment for streamlined detection and whole-genome sequencing of HCV and other highly diverse pathogens. F1000Research. 2015;4:1062. doi: 10.12688/f1000research.7111.1. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
