

Genetic basis and molecular profiling in myeloproliferative neoplasms
- PMID: 36347013
- PMCID: PMC10646774
- DOI: 10.1182/blood.2022017578
Genetic basis and molecular profiling in myeloproliferative neoplasms
Abstract
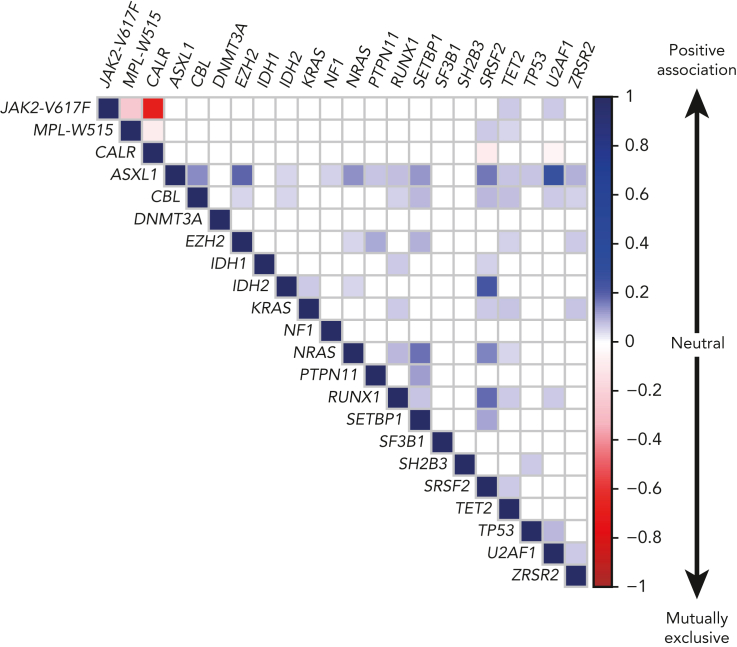
BCR::ABL1-negative myeloproliferative neoplasms (MPNs) are clonal diseases originating from a single hematopoietic stem cell that cause excessive production of mature blood cells. The 3 subtypes, that is, polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF), are diagnosed according to the World Health Organization (WHO) and international consensus classification (ICC) criteria. Acquired gain-of-function mutations in 1 of 3 disease driver genes (JAK2, CALR, and MPL) are the causative events that can alone initiate and promote MPN disease without requiring additional cooperating mutations. JAK2-p.V617F is present in >95% of PV patients, and also in about half of the patients with ET or PMF. ET and PMF are also caused by mutations in CALR or MPL. In ∼10% of MPN patients, those referred to as being "triple negative," none of the known driver gene mutations can be detected. The common theme between the 3 driver gene mutations and triple-negative MPN is that the Janus kinase-signal transducer and activator of transcription (JAK/STAT) signaling pathway is constitutively activated. We review the recent advances in our understanding of the early events after the acquisition of a driver gene mutation. The limiting factor that determines the frequency at which MPN disease develops with a long latency is not the acquisition of driver gene mutations, but rather the expansion of the clone. Factors that control the conversion from clonal hematopoiesis to MPN disease include inherited predisposition, presence of additional mutations, and inflammation. The full extent of knowledge of the mutational landscape in individual MPN patients is now increasingly being used to predict outcome and chose the optimal therapy.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict of interest disclosure: R.C.S. is a scientific advisory board member for and has equity in Ajax Therapeutics; and consulted for and received honoraria from Novartis and BMS/Celgene. R.K. is a scientific advisory board member for AOP Orphan Pharmaceuticals; and is an equity holder in/scientific advisor for MyeloPro D&R GmbH. D.L.P. declares no competing financial interests.
Figures




Comment in
-
Introduction to a review series on classic myeloproliferative neoplasms.Blood. 2023 Apr 20;141(16):1897-1899. doi: 10.1182/blood.2023019876. Blood. 2023. PMID: 36867843 No abstract available.
References
-
- Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 1976;295(17):913–916. - PubMed
-
- Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–679. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
