

The phenotypic landscape of essential human genes
- PMID: 36347254
- PMCID: PMC10482496
- DOI: 10.1016/j.cell.2022.10.017
The phenotypic landscape of essential human genes
Abstract
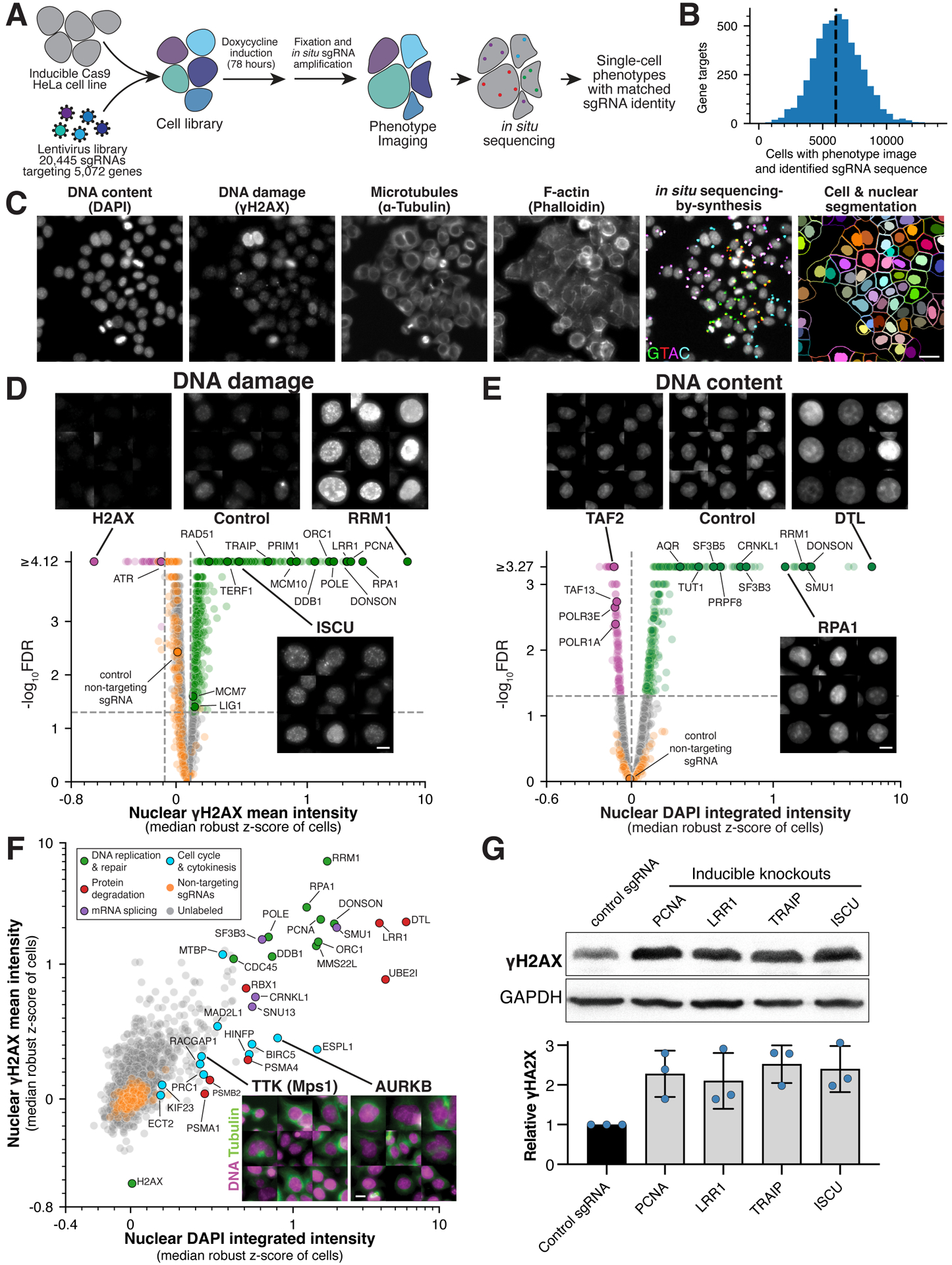
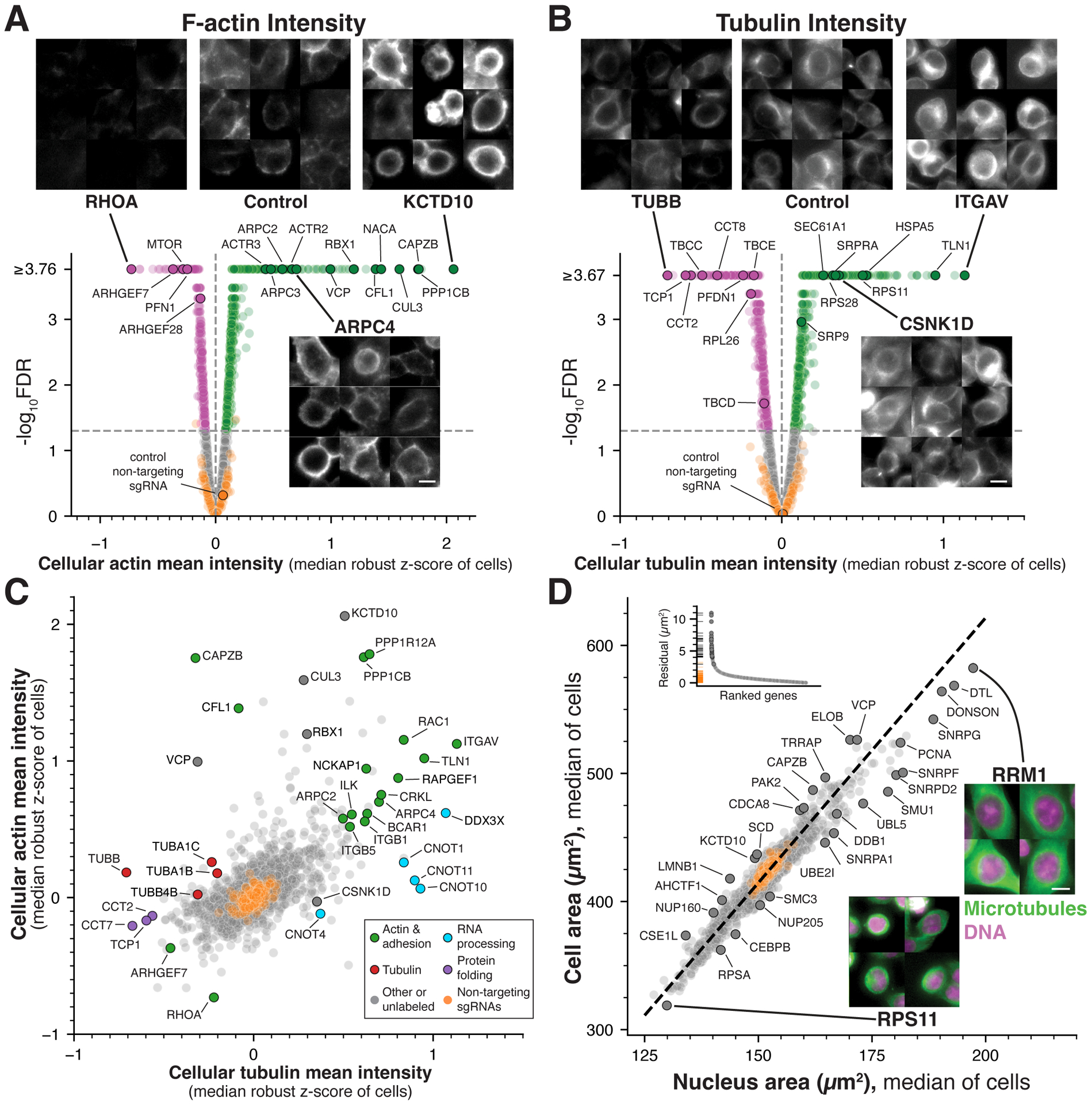
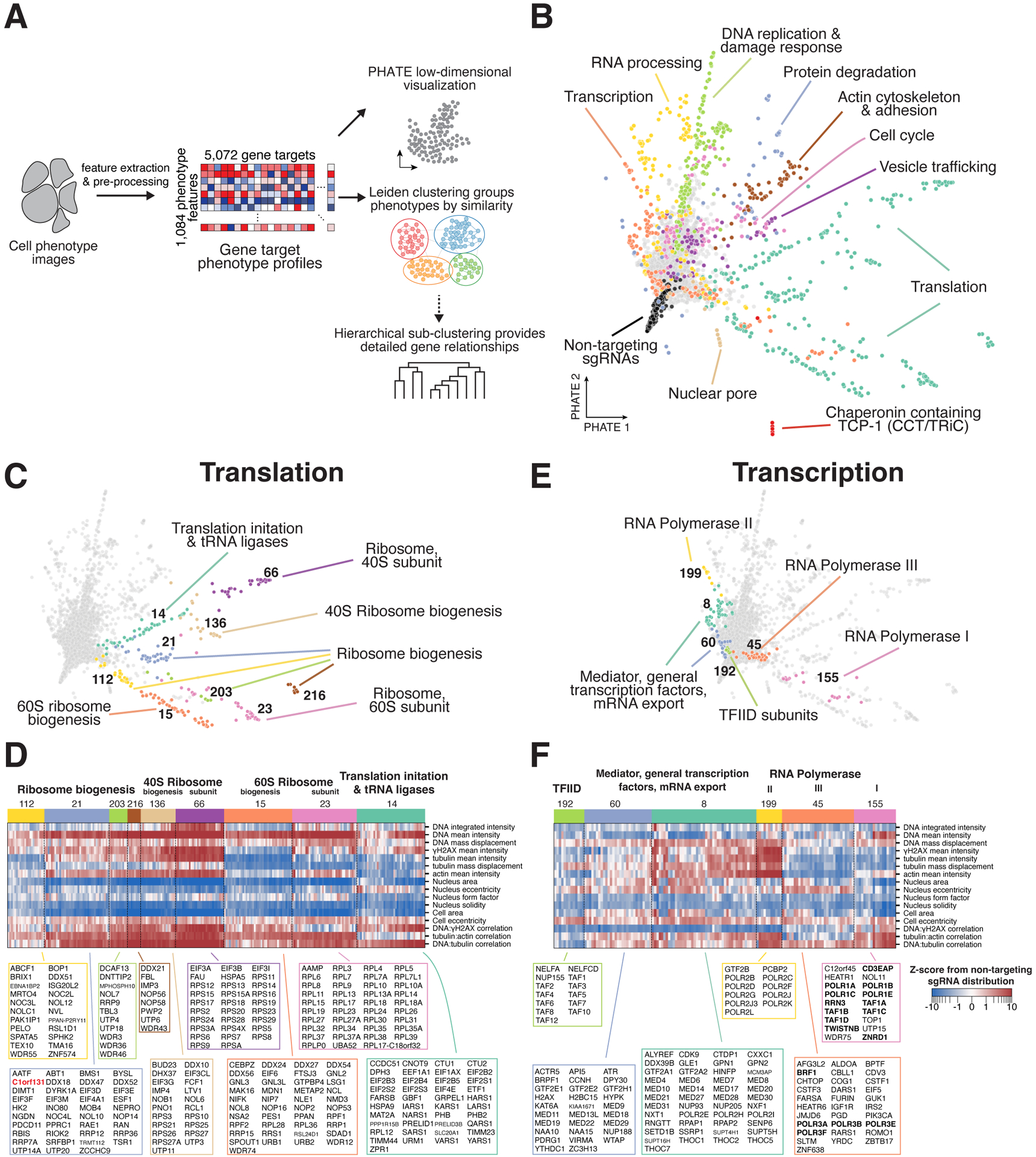
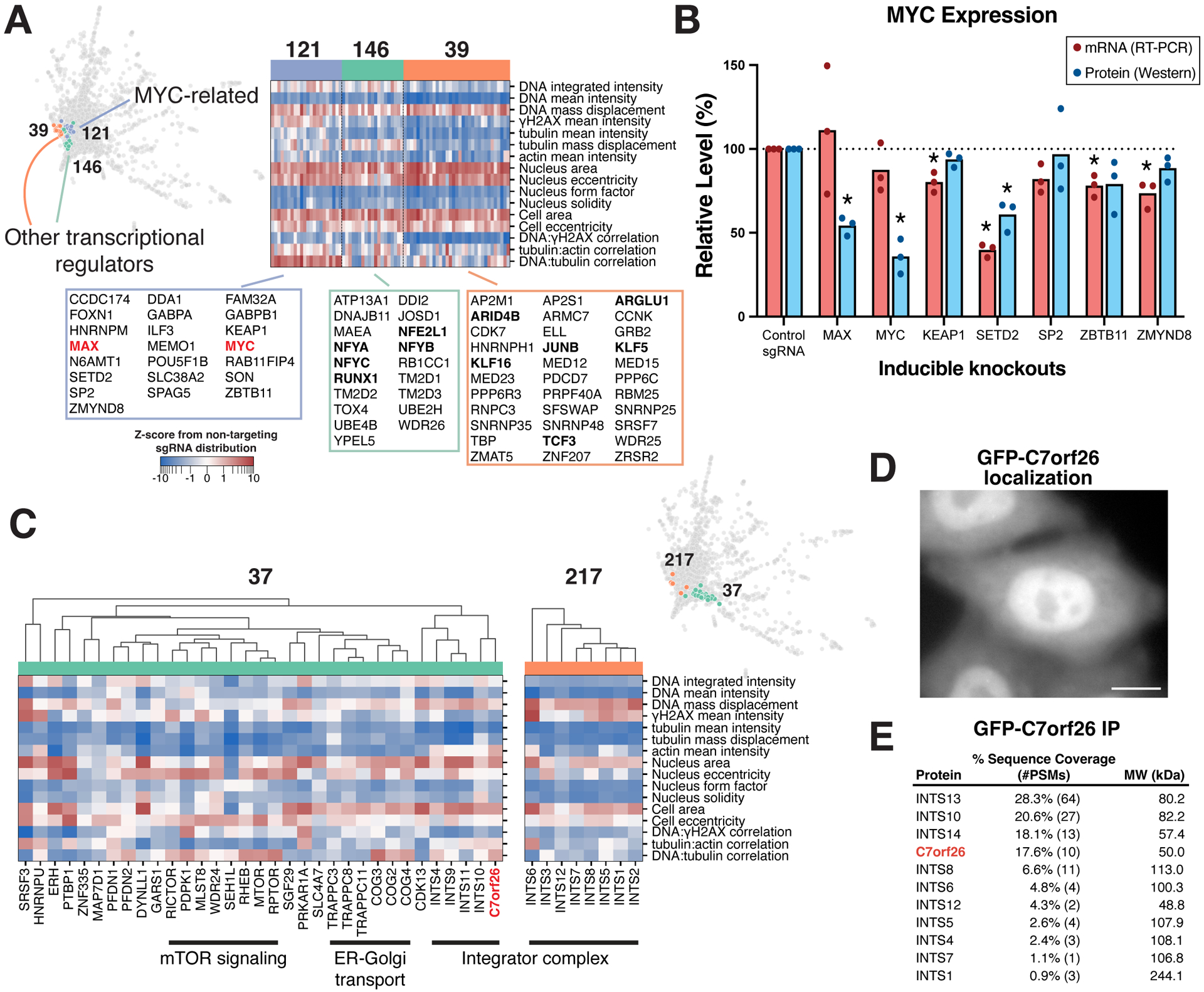
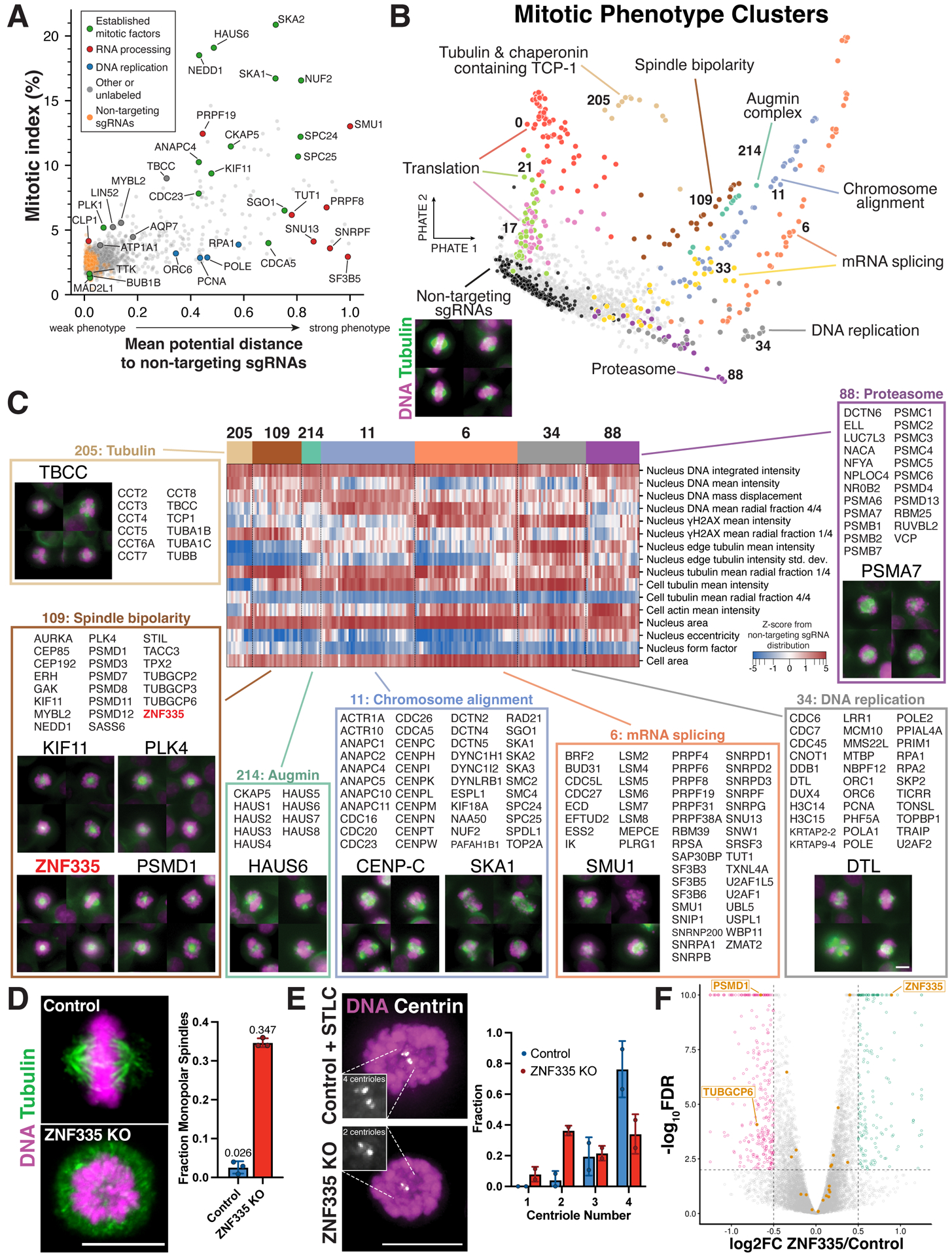
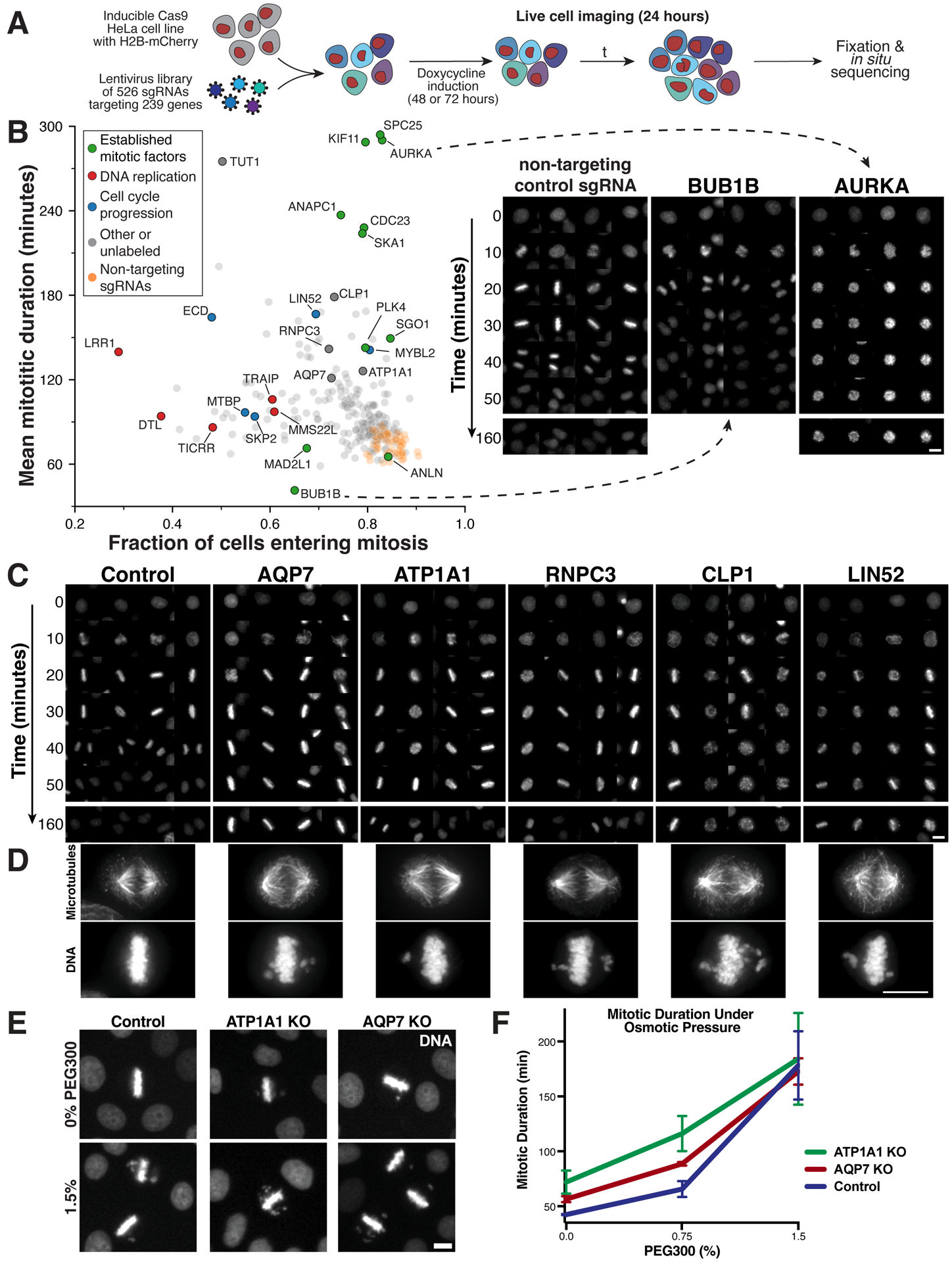
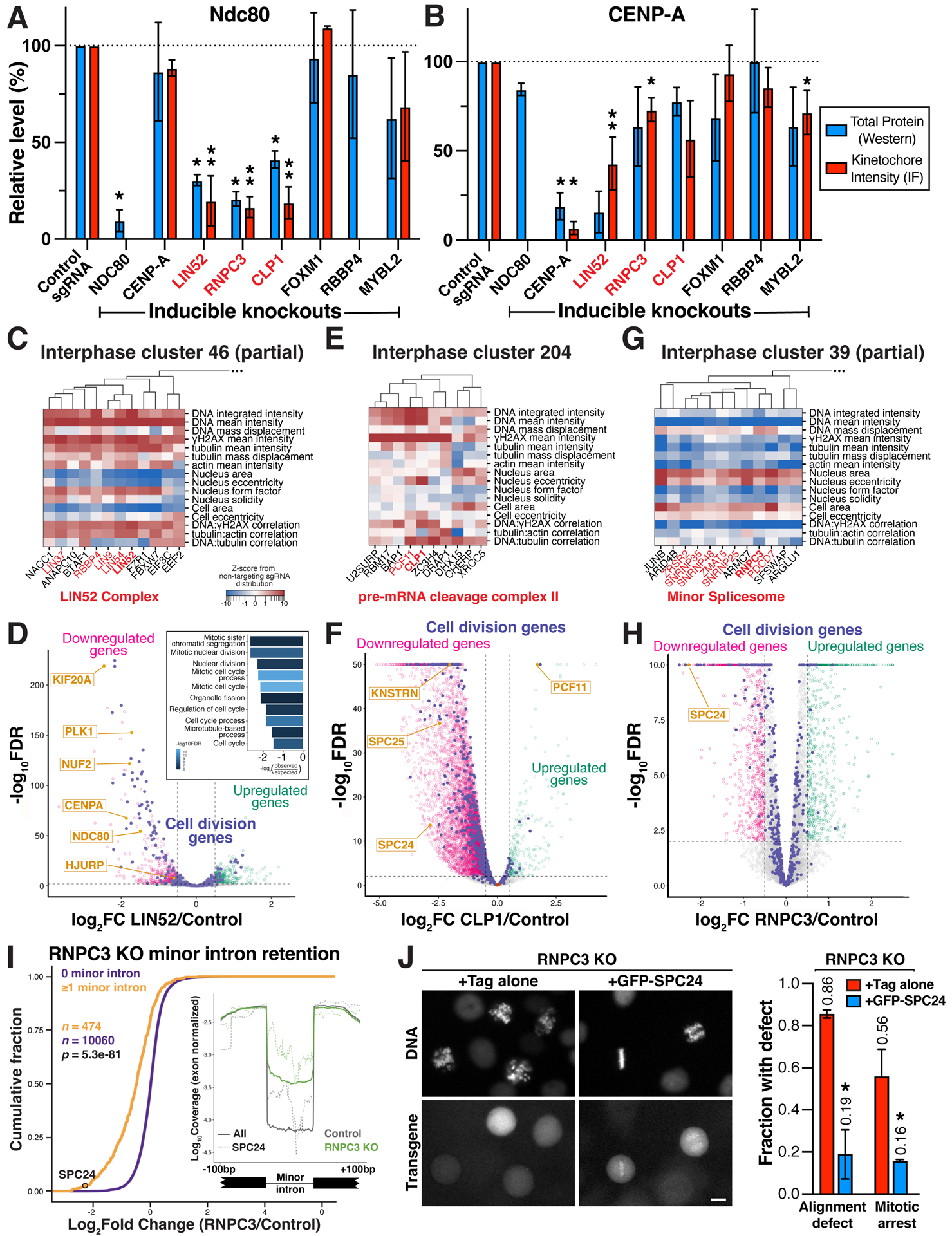
Understanding the basis for cellular growth, proliferation, and function requires determining the roles of essential genes in diverse cellular processes, including visualizing their contributions to cellular organization and morphology. Here, we combined pooled CRISPR-Cas9-based functional screening of 5,072 fitness-conferring genes in human HeLa cells with microscopy-based imaging of DNA, the DNA damage response, actin, and microtubules. Analysis of >31 million individual cells identified measurable phenotypes for >90% of gene knockouts, implicating gene targets in specific cellular processes. Clustering of phenotypic similarities based on hundreds of quantitative parameters further revealed co-functional genes across diverse cellular activities, providing predictions for gene functions and associations. By conducting pooled live-cell screening of ∼450,000 cell division events for 239 genes, we additionally identified diverse genes with functional contributions to chromosome segregation. Our work establishes a resource detailing the consequences of disrupting core cellular processes that represents the functional landscape of essential human genes.
Keywords: CRISPR-Cas9; cell division; essential genes; functional genomics; high-content screening; in situ sequencing; microscopy; mitosis; morphology; optical pooled screening.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests P.C.B. is a consultant to and/or equity holder in companies in the life sciences industries including 10X Genomics, GALT, Celsius Therapeutics, Next Generation Diagnostics, Cache DNA, Concerto Biosciences, and Stately. P.C.B.’s laboratory receives research funding from Calico Life Sciences and Merck for work related to genetic screening. The Broad Institute and MIT have filed U.S. patent applications on work described here and may seek to license the technology.
Figures
Comment in
-
Assigning phenotypes to essential human genes.Nat Rev Genet. 2023 Jan;24(1):2. doi: 10.1038/s41576-022-00558-6. Nat Rev Genet. 2023. PMID: 36414714 No abstract available.
References
-
- Condon KJ, Orozco JM, Adelmann CH, Spinelli JB, van der Helm PW, Roberts JM, Kunchok T, and Sabatini DM (2021). Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc. Natl. Acad. Sci. U. S. A 118, e2022120118. 10.1073/pnas.2022120118. - DOI - PMC - PubMed
-
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. 10.1016/j.cell.2016.11.038. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
