Probing the dynamic RNA structurome and its functions
- PMID: 36348050
- PMCID: PMC9644009
- DOI: 10.1038/s41576-022-00546-w
Probing the dynamic RNA structurome and its functions
Abstract
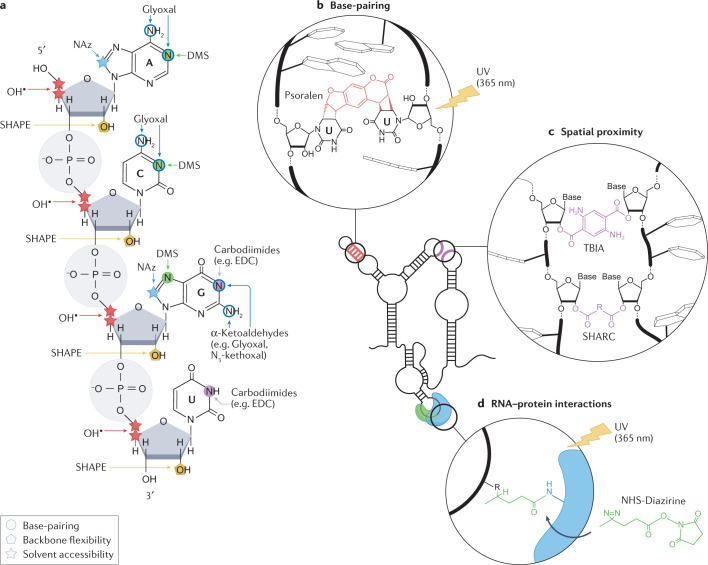
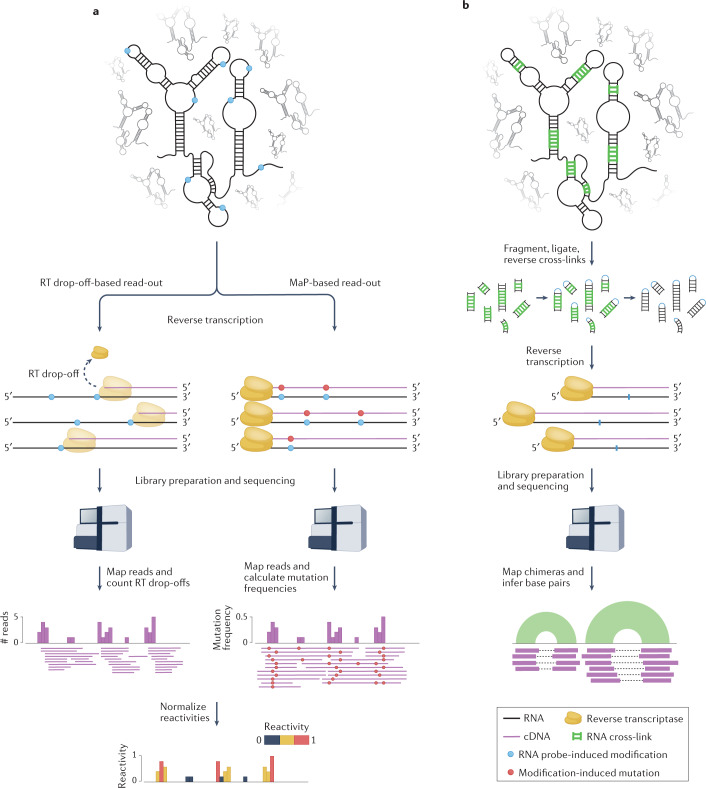
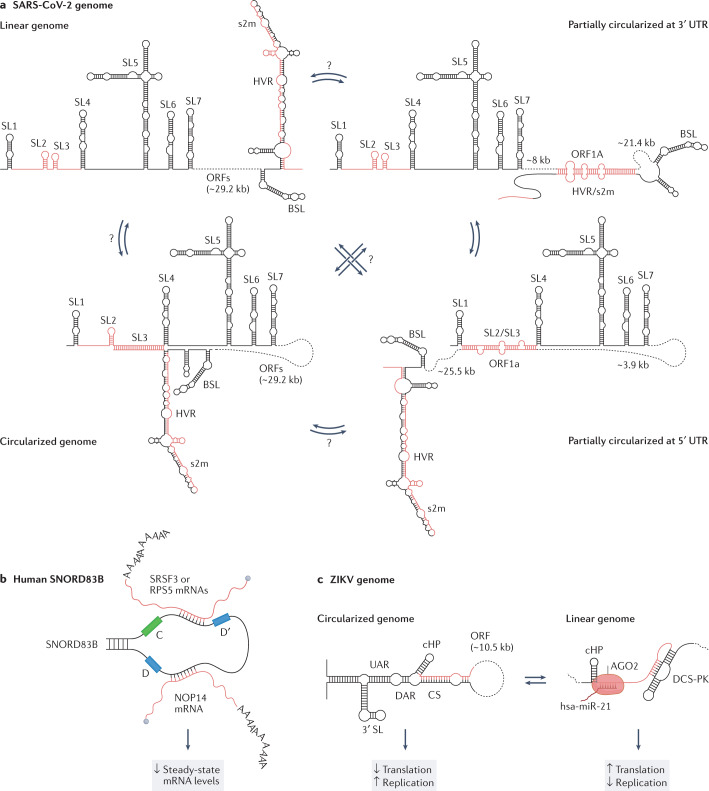
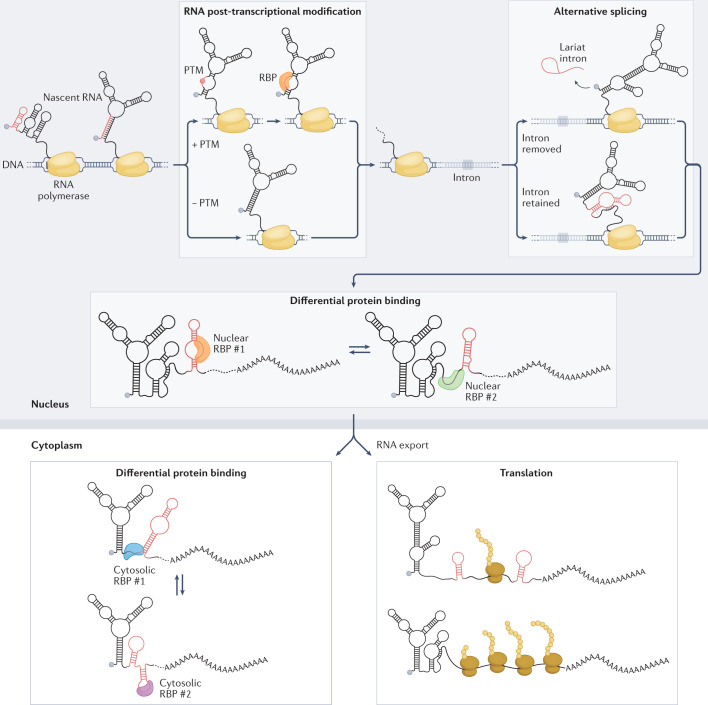
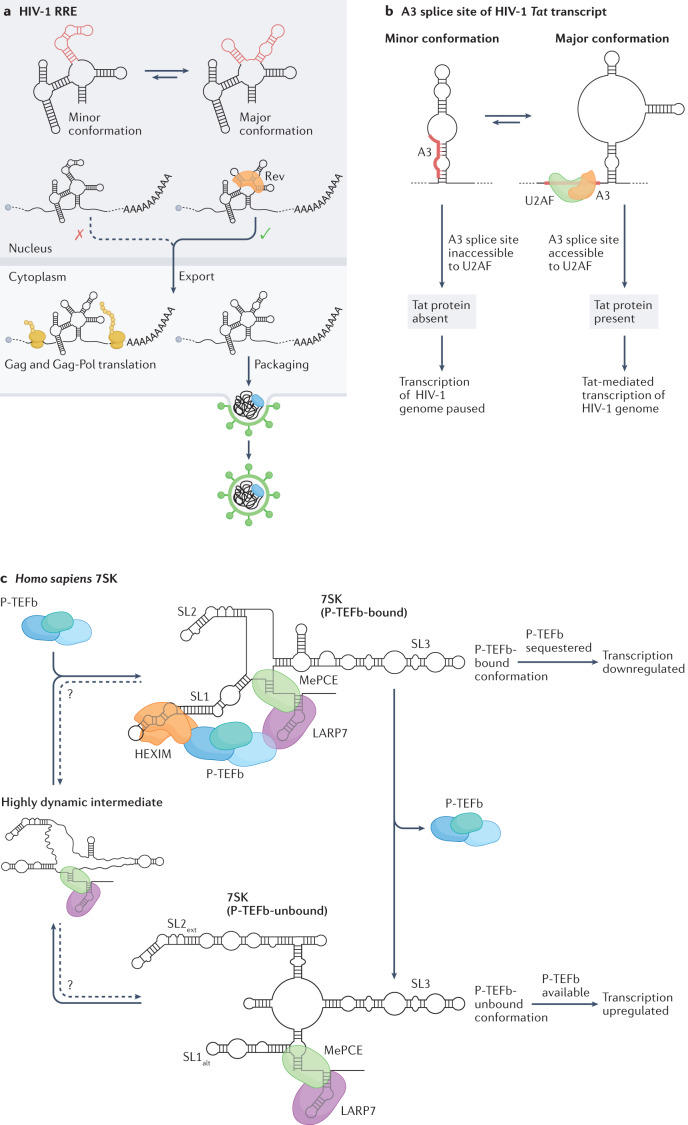
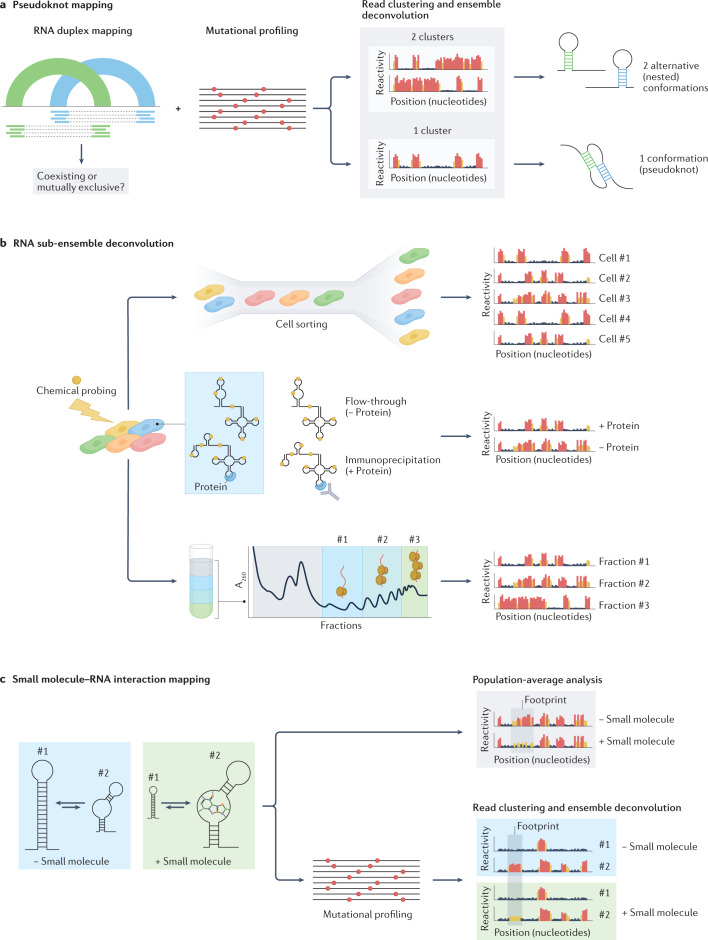
RNA is a key regulator of almost every cellular process, and the structures adopted by RNA molecules are thought to be central to their functions. The recent fast-paced evolution of high-throughput sequencing-based RNA structure mapping methods has enabled the rapid in vivo structural interrogation of entire cellular transcriptomes. Collectively, these studies are shedding new light on the long underestimated complexity of the structural organization of the transcriptome - the RNA structurome. Moreover, recent analyses are challenging the view that the RNA structurome is a static entity by revealing how RNA molecules establish intricate networks of alternative intramolecular and intermolecular interactions and that these ensembles of RNA structures are dynamically regulated to finely tune RNA functions in living cells. This new understanding of how RNA can shape cell phenotypes has important implications for the development of RNA-targeted therapeutic strategies.
© 2022. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures