

Enterovirus detection in different regions of Madagascar reveals a higher abundance of enteroviruses of species C in areas where several outbreaks of vaccine-derived polioviruses occurred
- PMID: 36348312
- PMCID: PMC9641760
- DOI: 10.1186/s12879-022-07826-0
Enterovirus detection in different regions of Madagascar reveals a higher abundance of enteroviruses of species C in areas where several outbreaks of vaccine-derived polioviruses occurred
Abstract
Background: Poliomyelitis outbreaks due to pathogenic vaccine-derived polioviruses (VDPVs) are threatening and complicating the global polio eradication initiative. Most of these VDPVs are genetic recombinants with non-polio enteroviruses (NPEVs) of species C. Little is known about factors favoring this genetic macroevolution process. Since 2001, Madagascar has experienced several outbreaks of poliomyelitis due to VDPVs, and most of VDPVs were isolated in the south of the island. The current study explored some of the viral factors that can promote and explain the emergence of recombinant VDPVs in Madagascar.
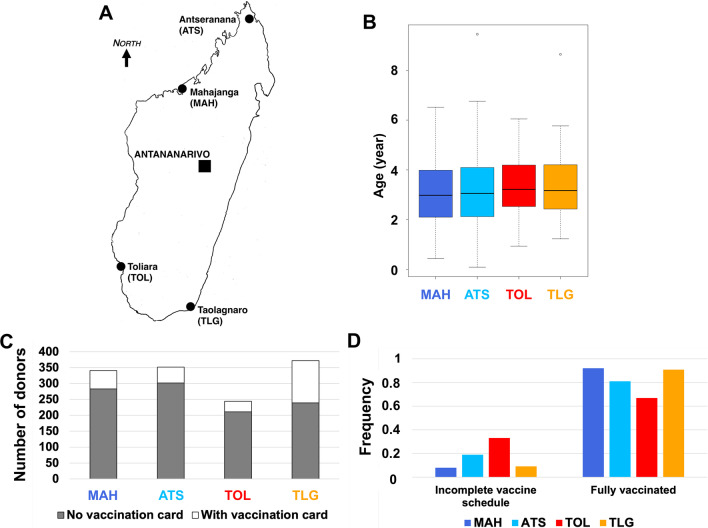
Methods: Between May to August 2011, we collected stools from healthy children living in two southern and two northern regions of Madagascar. Virus isolation was done in RD, HEp-2c, and L20B cell lines, and enteroviruses were detected using a wide-spectrum 5'-untranslated region RT-PCR assay. NPEVs were then sequenced for the VP1 gene used for viral genotyping.
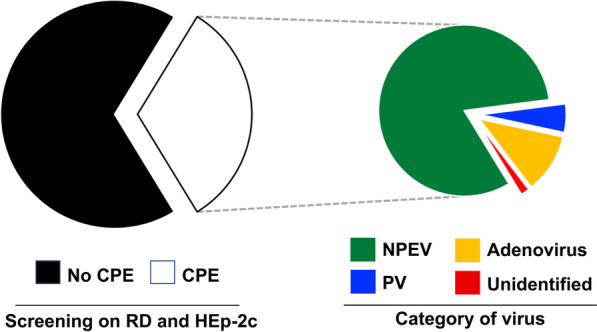
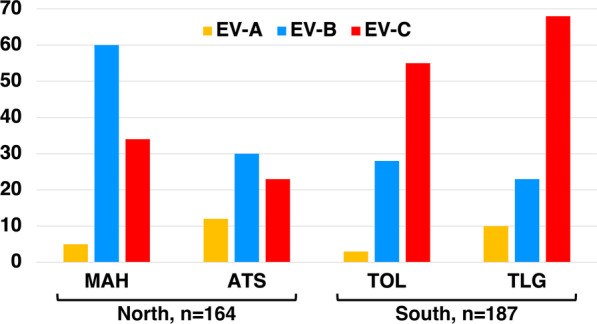
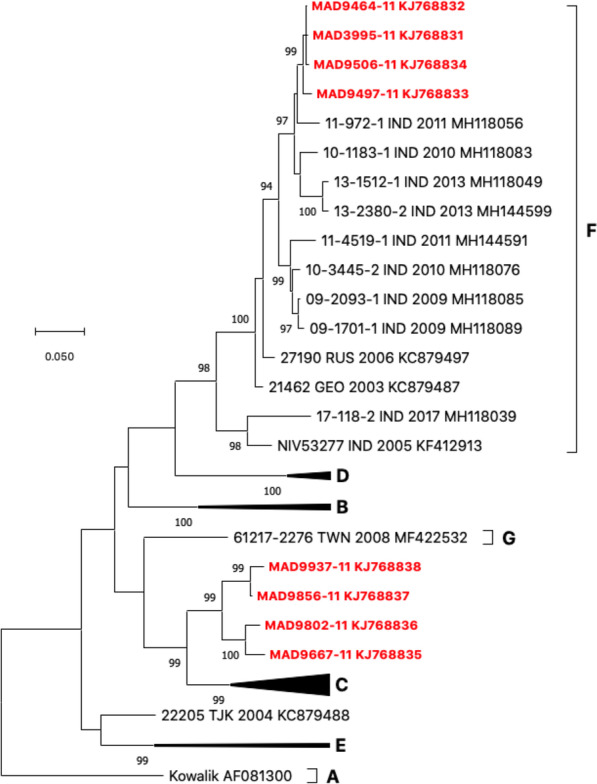
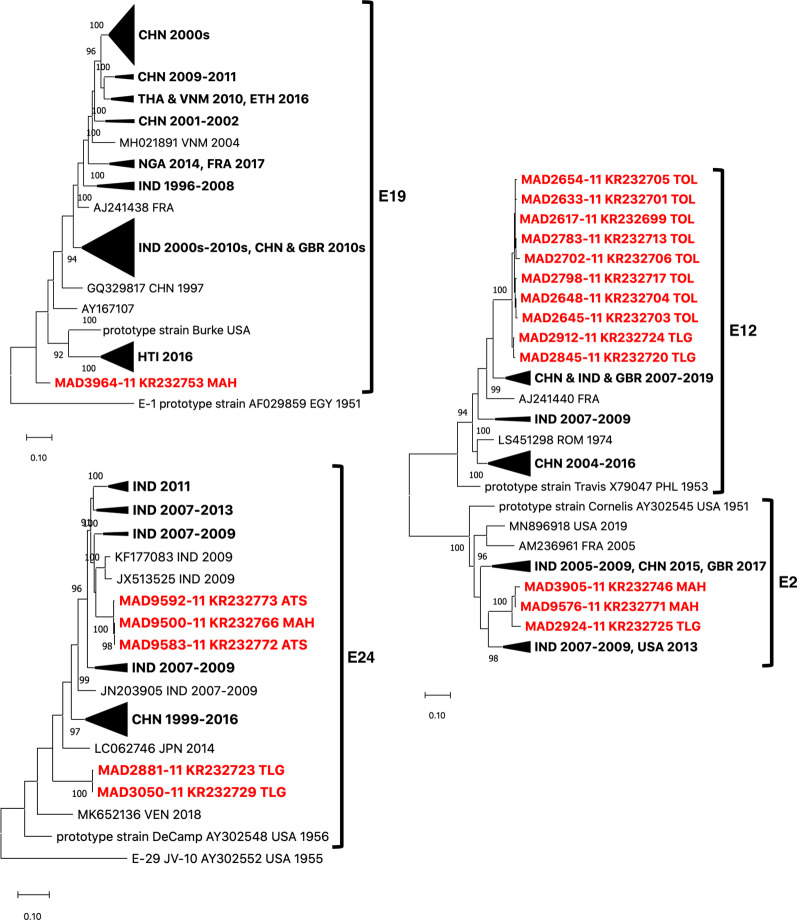
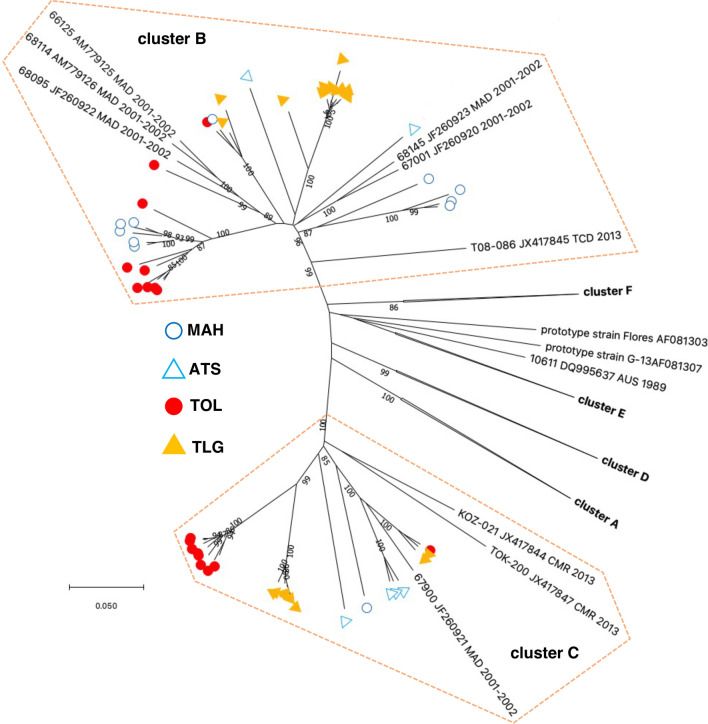
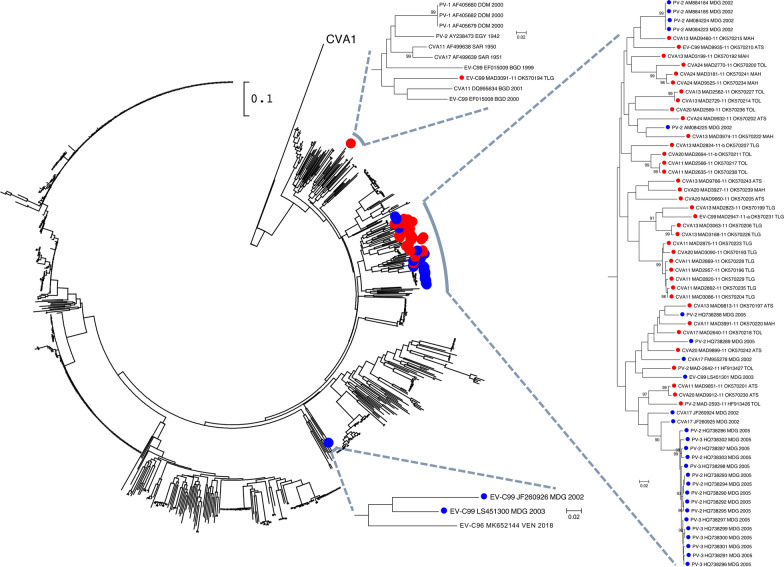
Results: Overall, we collected 1309 stools, of which 351 NPEVs (26.8%) were identified. Sequencing revealed 33 types of viruses belonging to three different species: Enterovirus A (8.5%), Enterovirus B (EV-B, 40.2%), and Enterovirus C (EV-C, 51.3%). EV-C species included coxsackievirus A13, A17, and A20 previously described as putative recombination partners for poliovirus vaccine strains. Interestingly, the isolation rate was higher among stools originating from the South (30.3% vs. 23.6%, p-value = 0.009). EV-C were predominant in southern sites (65.7%) while EV-B predominated in northern sites (54.9%). The factors that explain the relative abundance of EV-C in the South are still unknown.
Conclusions: Whatever its causes, the relative abundance of EV-C in the South of Madagascar may have promoted the infections of children by EV-C, including the PV vaccine strains, and have favored the recombination events between PVs and NPEVs in co-infected children, thus leading to the recurrent emergence of recombinant VDPVs in this region of Madagascar.
Keywords: Genotype; Human enterovirus; Madagascar; Vaccine-derived poliovirus.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no conflict of interests.
Figures
References
-
- Bao J, Thorley B, Isaacs D, Dinsmore N, Elliott EJ, McIntyre P, et al. Polio—The old foe and new challenges: an update for clinicians. J Paediatr Child Health. 2020;56:1527–1532. - PubMed
-
- Kew O, Pallansch M. Breaking the last chains of poliovirus transmission: progress and challenges in global polio eradication. Annu Rev Virol. 2018;5:427–451. - PubMed
-
- Burns CC, Diop OM, Sutter RW, Kew OM. Vaccine-derived polioviruses. J Infect Dis. 2014;210(Suppl 1):S283–293. - PubMed
-
- Rakoto-Andrianarivelo M, Gumede N, Jegouic S, Balanant J, Andriamamonjy SN, Rabemanantsoa S, et al. Reemergence of recombinant vaccine-derived poliovirus outbreak in Madagascar. J Infect Dis. 2008;197:1427–1435. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
