

Lineage abundance estimation for SARS-CoV-2 in wastewater using transcriptome quantification techniques
- PMID: 36348471
- PMCID: PMC9643916
- DOI: 10.1186/s13059-022-02805-9
Lineage abundance estimation for SARS-CoV-2 in wastewater using transcriptome quantification techniques
Abstract
Effectively monitoring the spread of SARS-CoV-2 mutants is essential to efforts to counter the ongoing pandemic. Predicting lineage abundance from wastewater, however, is technically challenging. We show that by sequencing SARS-CoV-2 RNA in wastewater and applying algorithms initially used for transcriptome quantification, we can estimate lineage abundance in wastewater samples. We find high variability in signal among individual samples, but the overall trends match those observed from sequencing clinical samples. Thus, while clinical sequencing remains a more sensitive technique for population surveillance, wastewater sequencing can be used to monitor trends in mutant prevalence in situations where clinical sequencing is unavailable.
© 2022. The Author(s).
Conflict of interest statement
N.D.G. is an infectious diseases consultant for Tempus Labs. W.P.H. is a scientific advisory board member to Biobot Analytics and has received compensation for expert witness testimony on the expected course of the pandemic. N.G. is a co-founder of Biobot Analytics; C.D., K.A.M., and M.I. are employees of Biobot Analytics.
Figures

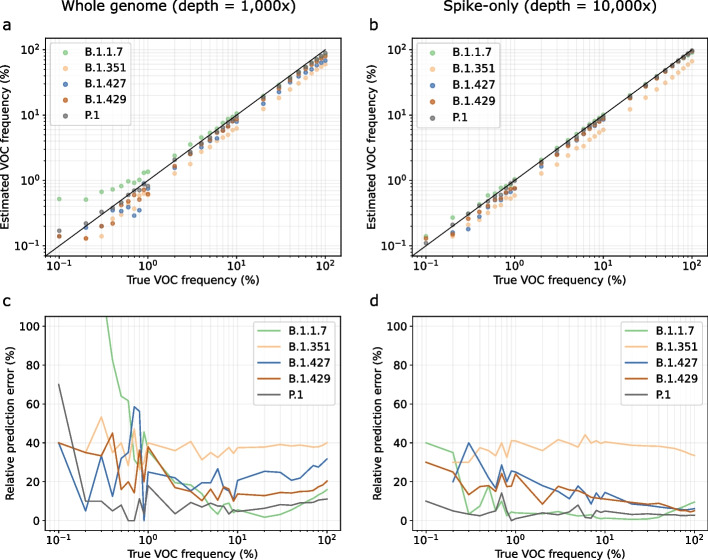


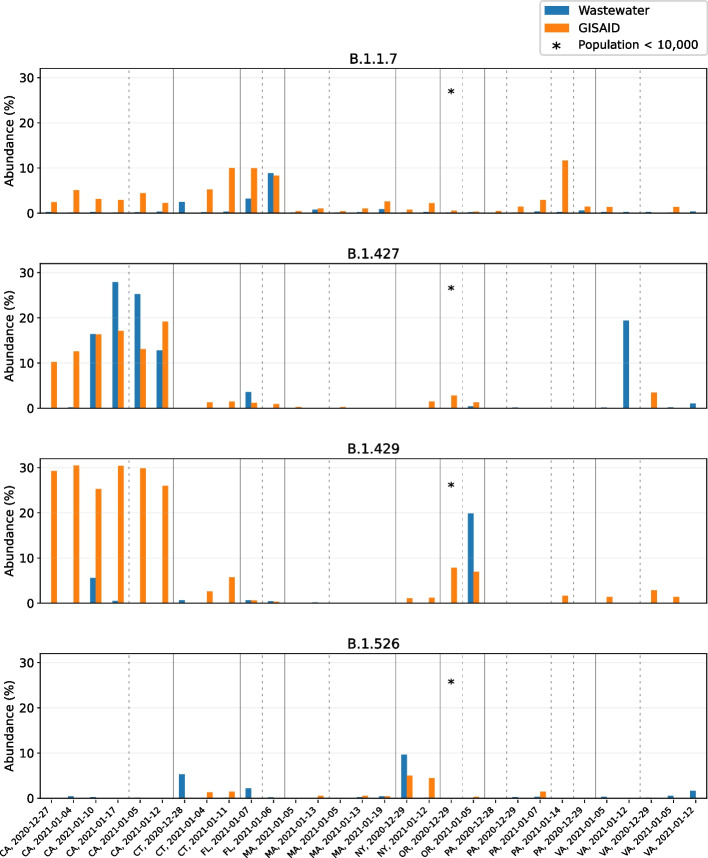
Update of
-
Variant abundance estimation for SARS-CoV-2 in wastewater using RNA-Seq quantification.medRxiv [Preprint]. 2021 Sep 2:2021.08.31.21262938. doi: 10.1101/2021.08.31.21262938. medRxiv. 2021. Update in: Genome Biol. 2022 Nov 8;23(1):236. doi: 10.1186/s13059-022-02805-9. PMID: 34494031 Free PMC article. Updated. Preprint.
References
-
- Knyazev S, et al. Unlocking capacities of viral genomics for the COVID-19 pandemic response. 2021.
-
- CDC . SARS-CoV-2 Variant Classifications and Definitions. 2021.
-
- GISAID - Initiative. https://www.gisaid.org/.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
