

Good response to the late treatment with ataluren in a boy with Duchenne muscular dystrophy: could the previous mild course of the disease have affected the outcome?
- PMID: 36349184
- PMCID: PMC9628801
- DOI: 10.36185/2532-1900-078
Good response to the late treatment with ataluren in a boy with Duchenne muscular dystrophy: could the previous mild course of the disease have affected the outcome?
Abstract
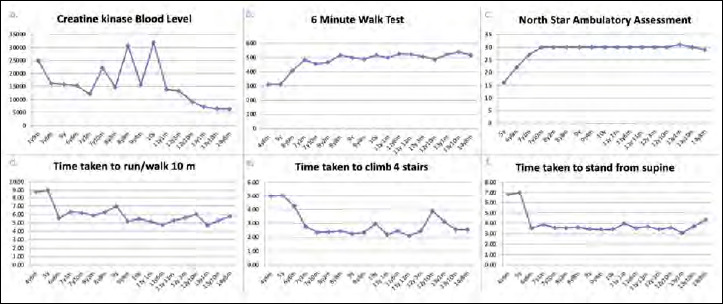
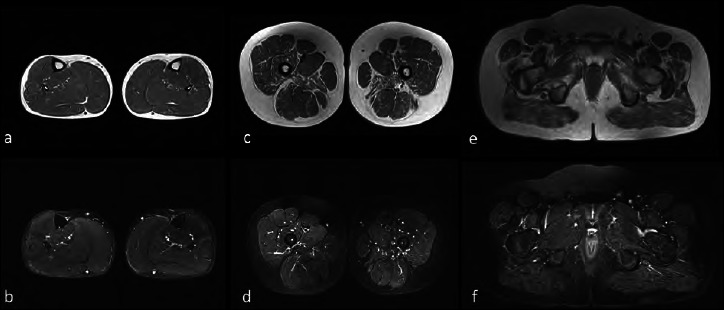
Duchenne muscular dystrophy (DMD) is a severe, progressive X-linked recessive disorder, caused by the absence of the dystrophin protein. A resolutive therapy for DMD is not yet available. The first approved drug for DMD patients with nonsense mutations is ataluren, approved for the treatment of children aged ≥ 2 yrs, that seems effective in slowing the disease progression. An earlier introduction of ataluren seems to give better results. We report the case of a 14-year-old DMD patient with a nonsense mutation in exon 70, still ambulant, who started taking ataluren at 12 years and remained stable for the following two years. The patient was on steroid since the age of 6, with beneficial effects. At two-years follow-up, an optimal disease evolution was observed, associated with a constant decrease of creatine kinase blood levels. Despite the late start of the treatment, ataluren seems to have significantly contributed to the stabilization of the functional status in this patient though it cannot be excluded that the result may have been influenced by the previous favorable course of the disease. However, further studies should be planned in patients with similar age treated with ataluren to better evaluate the treatment's results compared to the natural course of the disease.
Keywords: Duchenne muscular dystrophy; ataluren; nonsense mutation; target therapy; time function tests.
©2022 Gaetano Conte Academy - Mediterranean Society of Myology, Naples, Italy.
Figures
References
-
- Bushby K, Finkel R, Birnkrant DJ, et al. . Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77-93. https://doi.org/10.1016/S1474-4422(09)70271-6 10.1016/S1474-4422(09)70271-6 - DOI - PubMed
-
- Birnkfrant DJ, Bushby K, Bann C, et al. . Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018;17:347-361. https://doi.org/10.1016/S1474-4422(18)30025-5 10.1016/S1474-4422(18)30025-5 - DOI - PMC - PubMed
-
- Finkel RS, Flanigan KM, Wong B, et al. . Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS One 2013;8:e81302. https://doi.org/10.1371/journal.pone.0081302 10.1371/journal.pone.0081302 - DOI - PMC - PubMed
-
- Bushby K, Finkel R, Wong B, et al. . Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014:50:477-487. https://doi.org/10.1002/mus.24332 10.1002/mus.24332 - DOI - PMC - PubMed
-
- McDonald CM, Campbell C, Torricelli RE, et al. . Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;390:1489-1498. https://doi.org/10.1016/S0140-6736(17)31611-2 10.1016/S0140-6736(17)31611-2 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources