

Reversible Myc hypomorphism identifies a key Myc-dependency in early cancer evolution
- PMID: 36351945
- PMCID: PMC9646778
- DOI: 10.1038/s41467-022-34079-x
Reversible Myc hypomorphism identifies a key Myc-dependency in early cancer evolution
Abstract
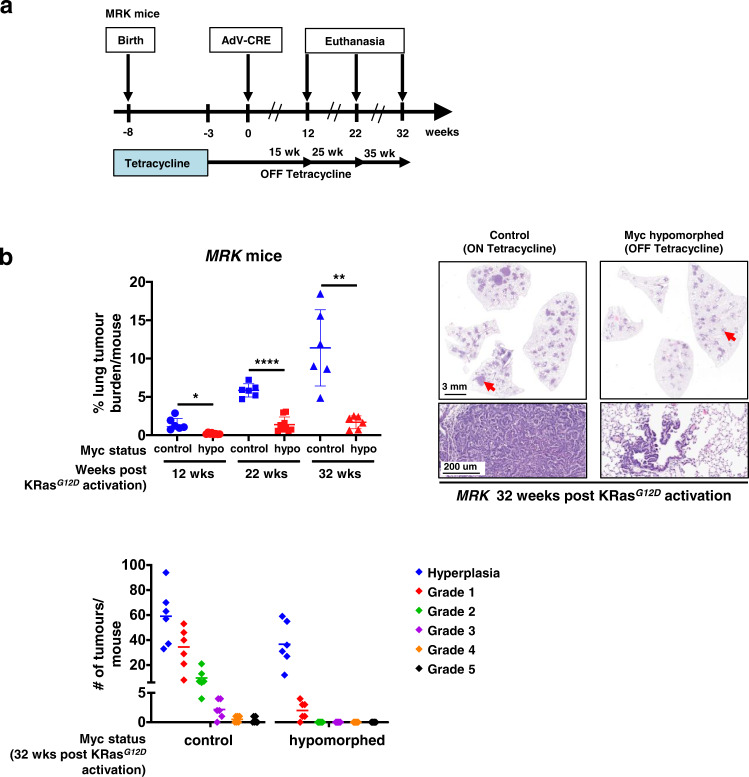
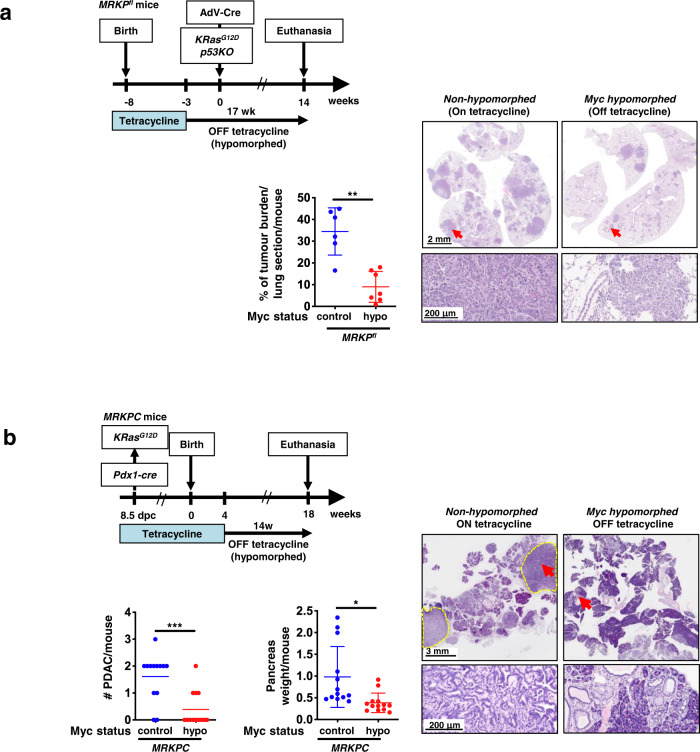
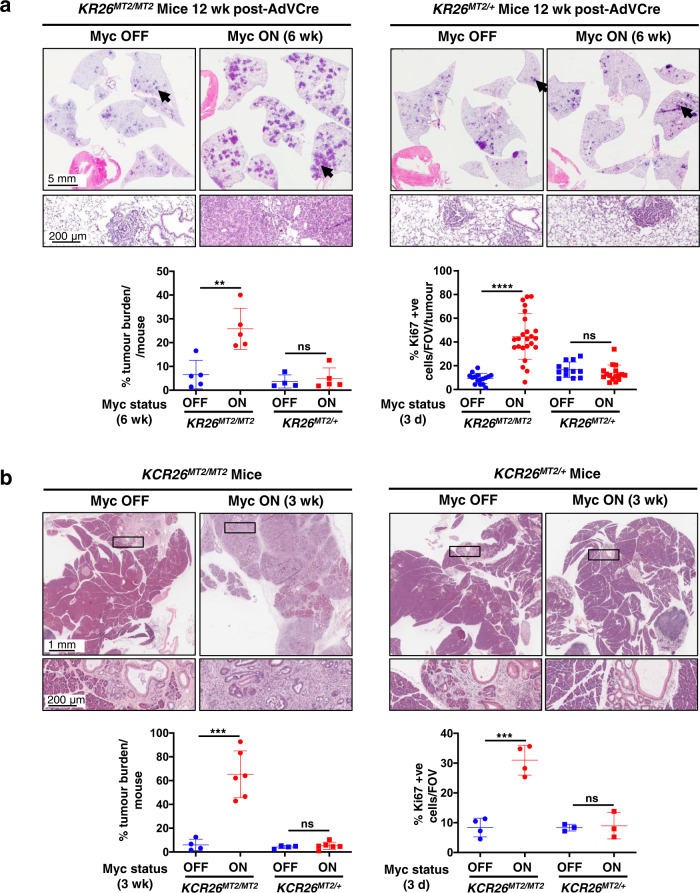
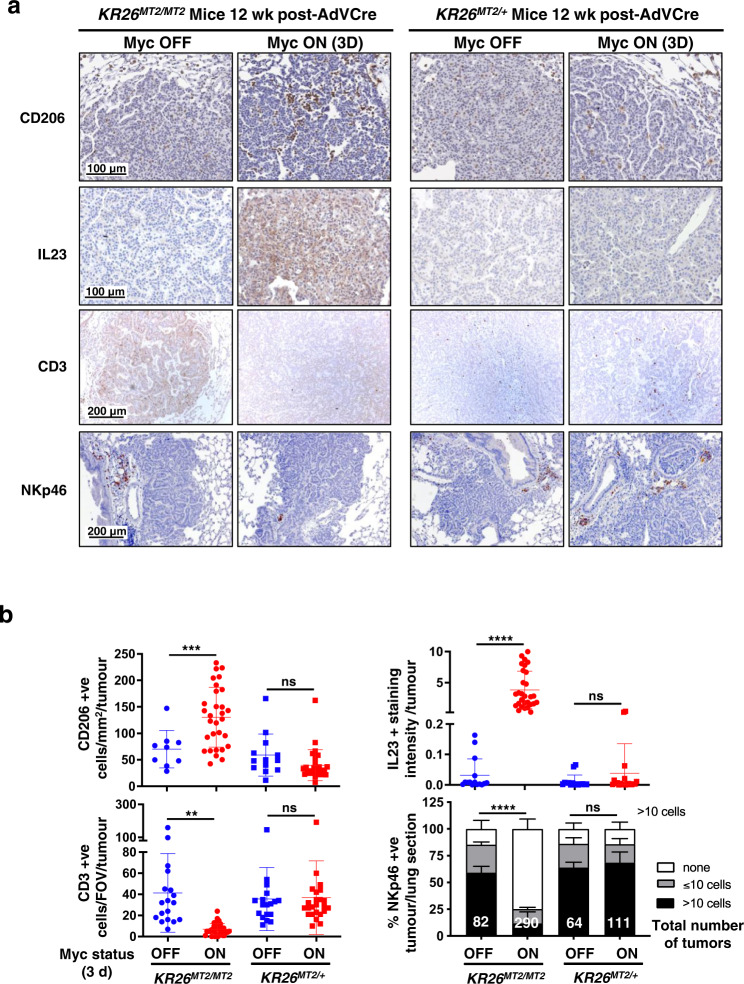
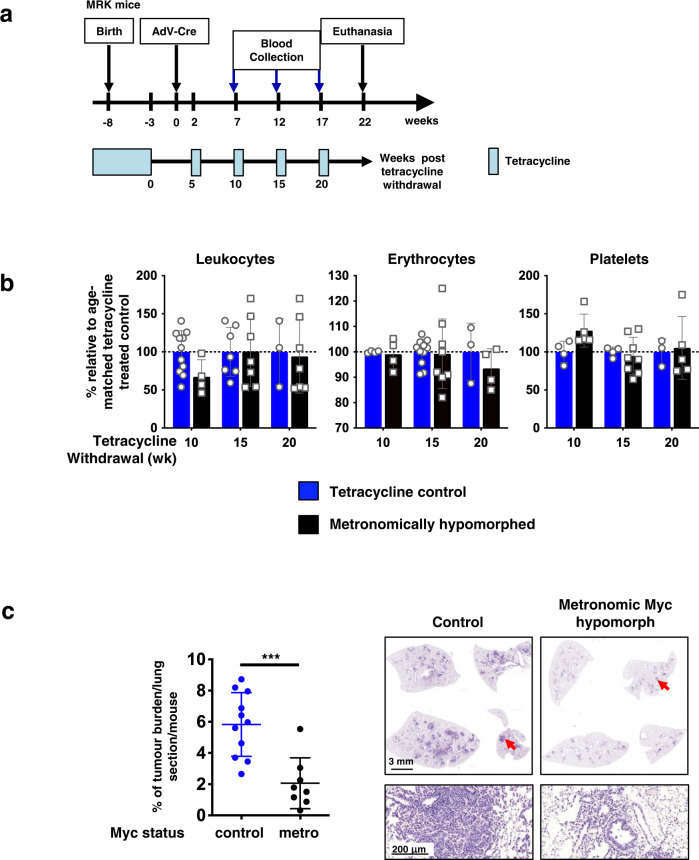
Germ-line hypomorphism of the pleiotropic transcription factor Myc in mice, either through Myc gene haploinsufficiency or deletion of Myc enhancers, delays onset of various cancers while mice remain viable and exhibit only relatively mild pathologies. Using a genetically engineered mouse model in which Myc expression may be systemically and reversibly hypomorphed at will, we asked whether this resistance to tumour progression is also emplaced when Myc hypomorphism is acutely imposed in adult mice. Indeed, adult Myc hypomorphism profoundly blocked KRasG12D-driven lung and pancreatic cancers, arresting their evolution at the early transition from indolent pre-tumour to invasive cancer. We show that such arrest is due to the incapacity of hypomorphic levels of Myc to drive release of signals that instruct the microenvironmental remodelling necessary to support invasive cancer. The cancer protection afforded by long-term adult imposition of Myc hypomorphism is accompanied by only mild collateral side effects, principally in haematopoiesis, but even these are circumvented if Myc hypomorphism is imposed metronomically whereas potent cancer protection is retained.
© 2022. The Author(s).
Conflict of interest statement
G.I.E. is a member of AstraZeneca’s IMED oncology external science advisory panel. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
