

Spatial genomics maps the structure, nature and evolution of cancer clones
- PMID: 36352222
- PMCID: PMC9668746
- DOI: 10.1038/s41586-022-05425-2
Spatial genomics maps the structure, nature and evolution of cancer clones
Abstract
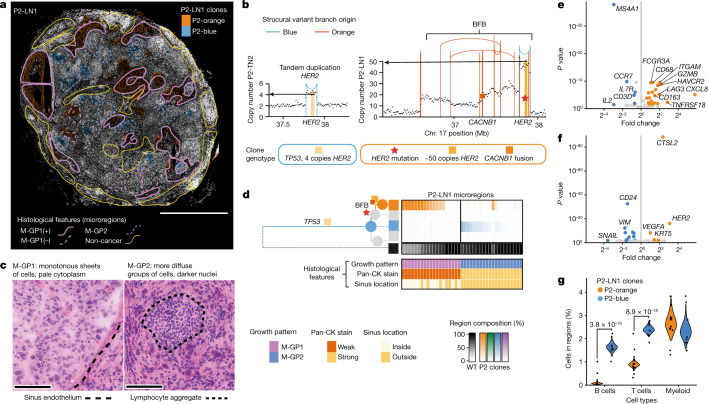
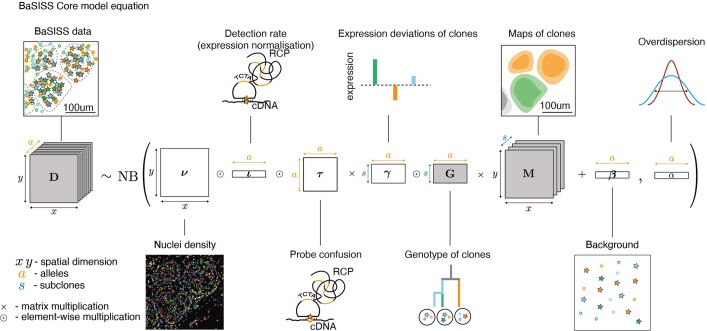
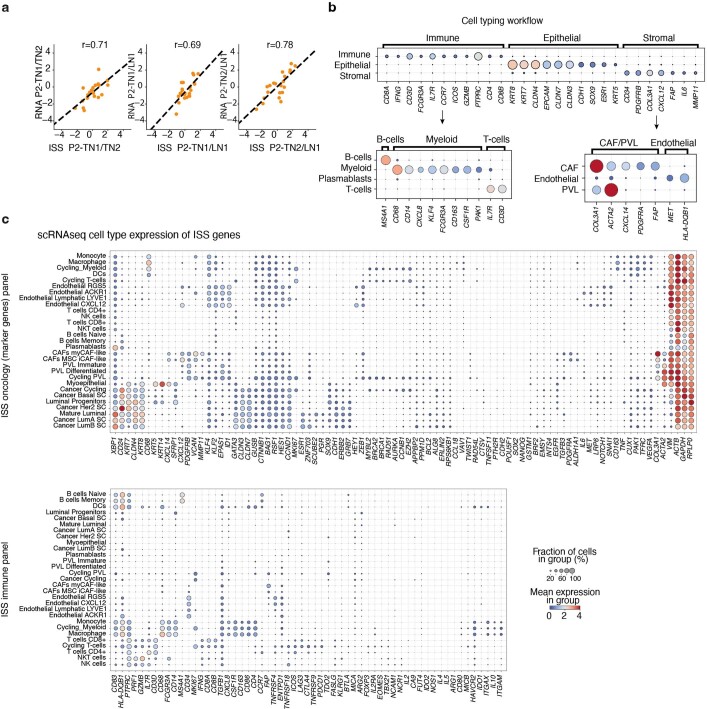
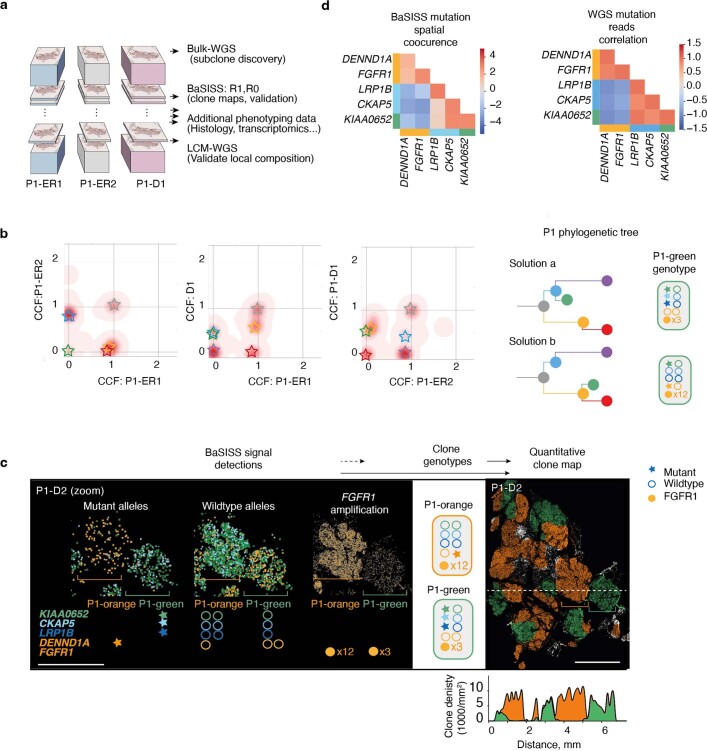
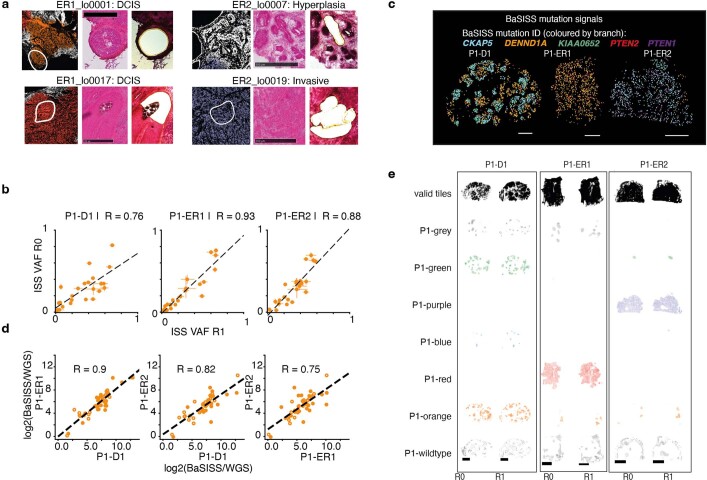
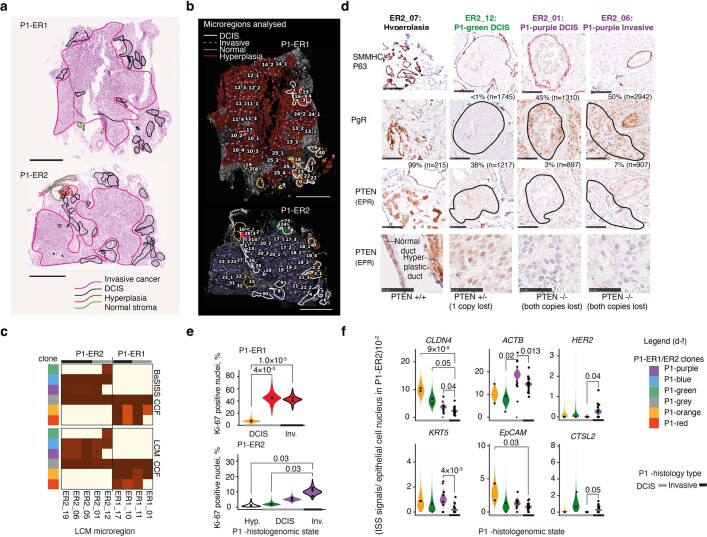
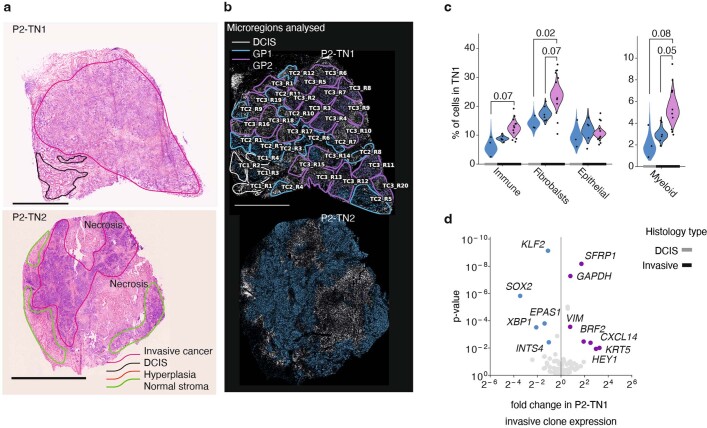
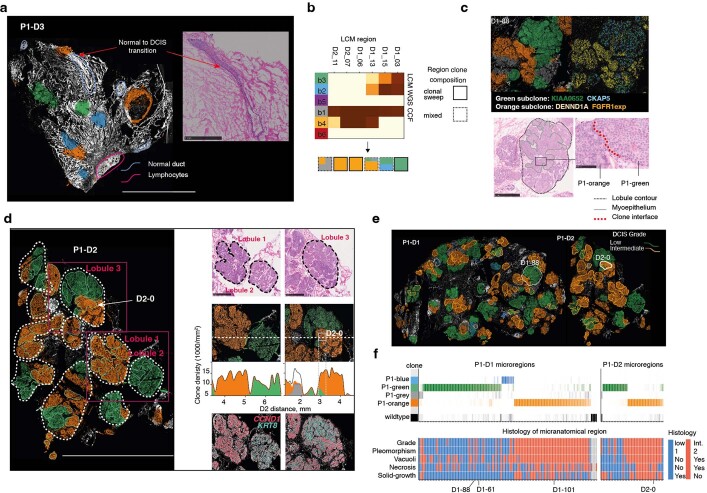
Genome sequencing of cancers often reveals mosaics of different subclones present in the same tumour1-3. Although these are believed to arise according to the principles of somatic evolution, the exact spatial growth patterns and underlying mechanisms remain elusive4,5. Here, to address this need, we developed a workflow that generates detailed quantitative maps of genetic subclone composition across whole-tumour sections. These provide the basis for studying clonal growth patterns, and the histological characteristics, microanatomy and microenvironmental composition of each clone. The approach rests on whole-genome sequencing, followed by highly multiplexed base-specific in situ sequencing, single-cell resolved transcriptomics and dedicated algorithms to link these layers. Applying the base-specific in situ sequencing workflow to eight tissue sections from two multifocal primary breast cancers revealed intricate subclonal growth patterns that were validated by microdissection. In a case of ductal carcinoma in situ, polyclonal neoplastic expansions occurred at the macroscopic scale but segregated within microanatomical structures. Across the stages of ductal carcinoma in situ, invasive cancer and lymph node metastasis, subclone territories are shown to exhibit distinct transcriptional and histological features and cellular microenvironments. These results provide examples of the benefits afforded by spatial genomics for deciphering the mechanisms underlying cancer evolution and microenvironmental ecology.
© 2022. The Author(s).
Conflict of interest statement
J.S. is now (but was not at the time of contribution to this manuscript) an employee of Spatial Transcriptomics, Part of 10X Genomics, Inc. Y.S.J. is co-founder of Genome Insight. C.S. is co-owner of HistoOne AB, and has research contracts with Prelude Dx. M.N. is an advisor to 10X Genomics. All other authors declare no competing interests.
Figures
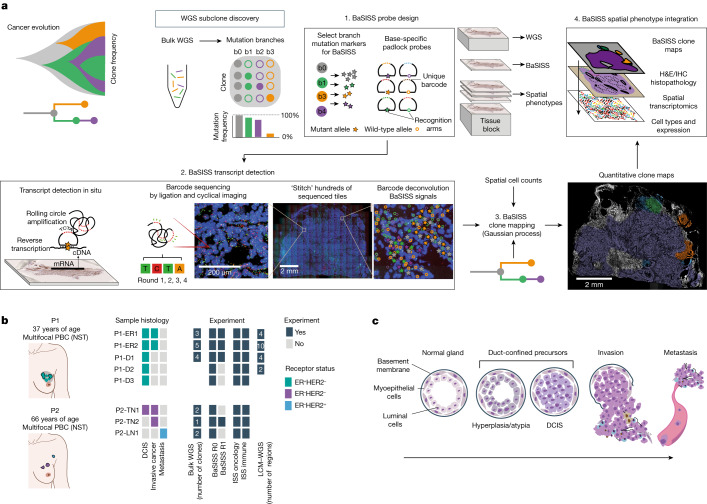
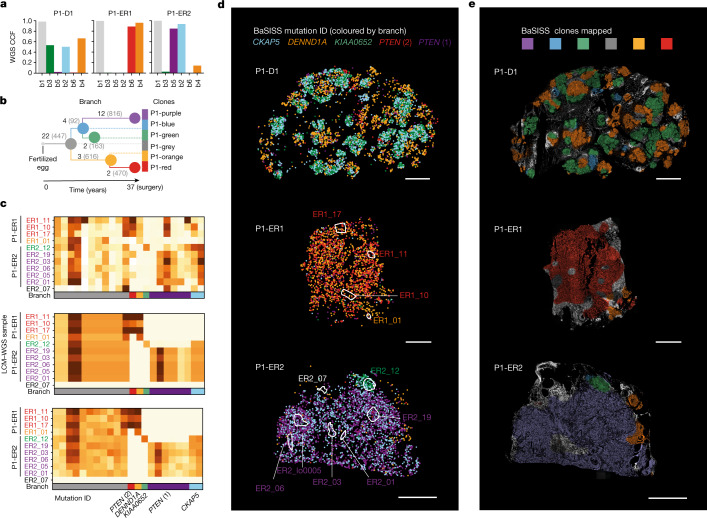
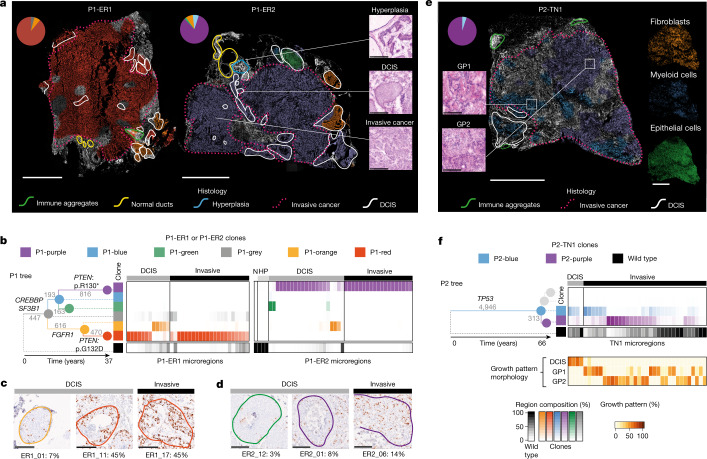
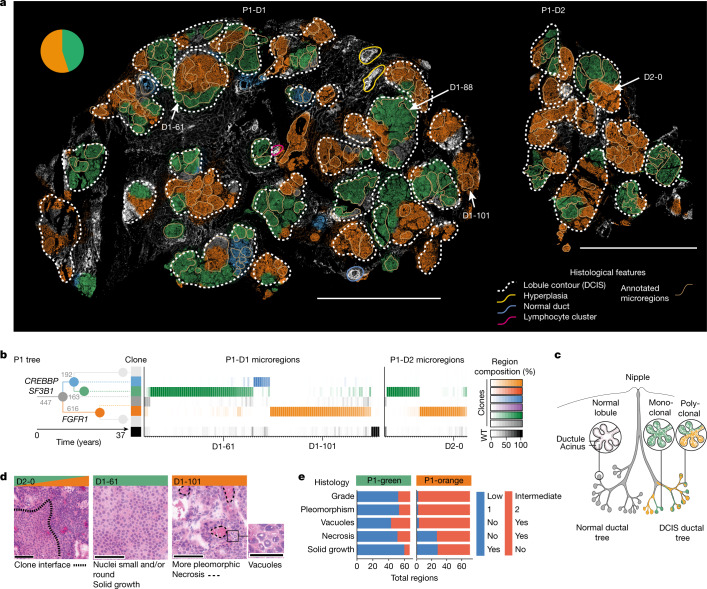


Comment in
-
Spatial maps of genetically diverse breast cancer cells.Nature. 2022 Nov;611(7936):458-459. doi: 10.1038/d41586-022-03560-4. Nature. 2022. PMID: 36352119 No abstract available.
References
-
- McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–628. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
