

Temporal change in chromatin accessibility predicts regulators of nodulation in Medicago truncatula
- PMID: 36352404
- PMCID: PMC9647978
- DOI: 10.1186/s12915-022-01450-9
Temporal change in chromatin accessibility predicts regulators of nodulation in Medicago truncatula
Abstract
Background: Symbiotic associations between bacteria and leguminous plants lead to the formation of root nodules that fix nitrogen needed for sustainable agricultural systems. Symbiosis triggers extensive genome and transcriptome remodeling in the plant, yet an integrated understanding of the extent of chromatin changes and transcriptional networks that functionally regulate gene expression associated with symbiosis remains poorly understood. In particular, analyses of early temporal events driving this symbiosis have only captured correlative relationships between regulators and targets at mRNA level. Here, we characterize changes in transcriptome and chromatin accessibility in the model legume Medicago truncatula, in response to rhizobial signals that trigger the formation of root nodules.
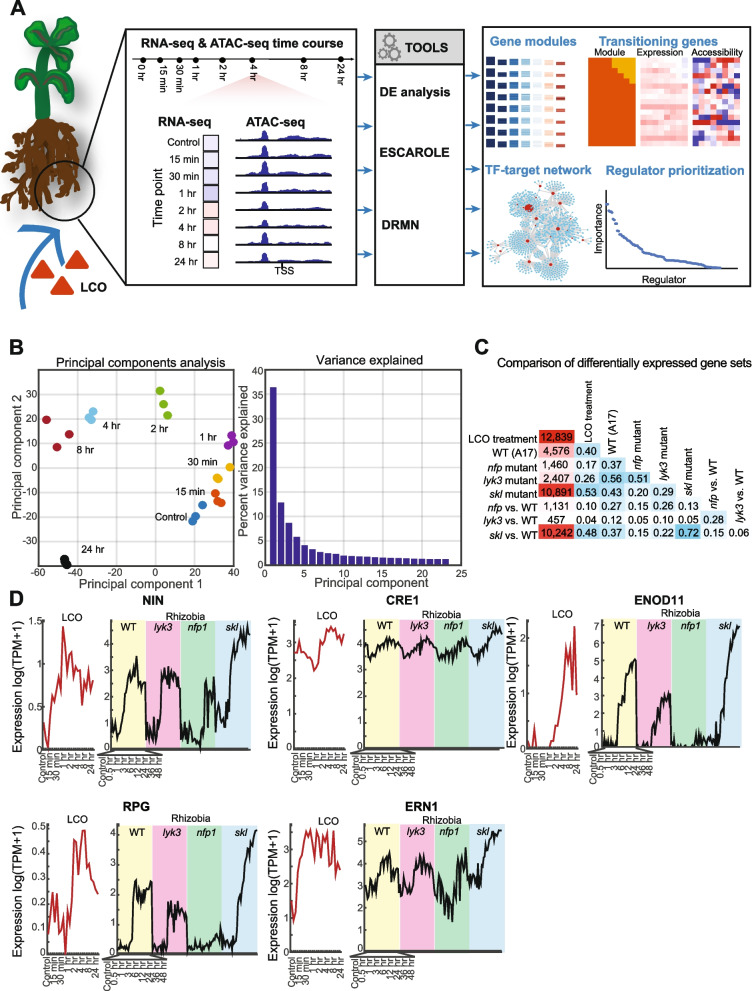
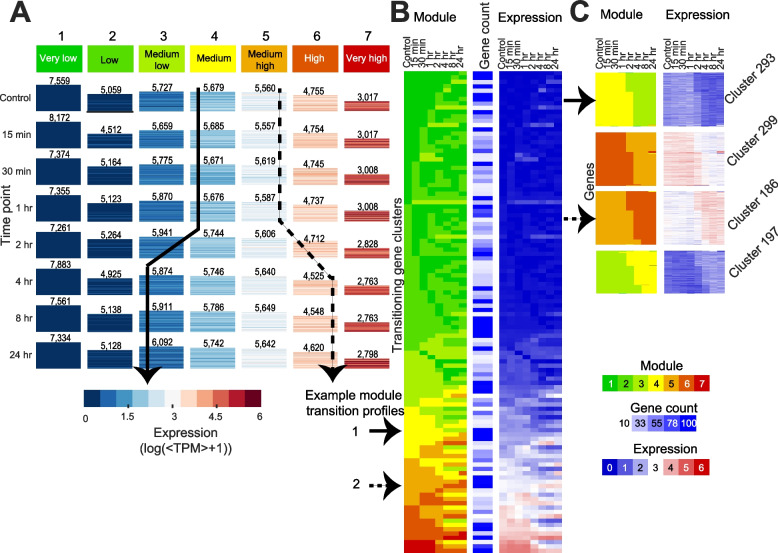
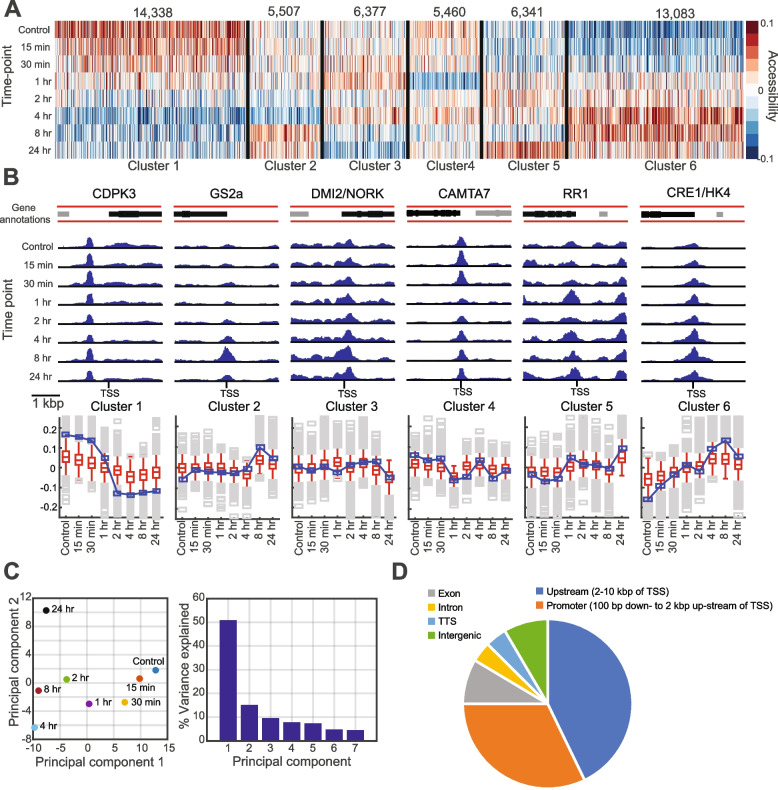
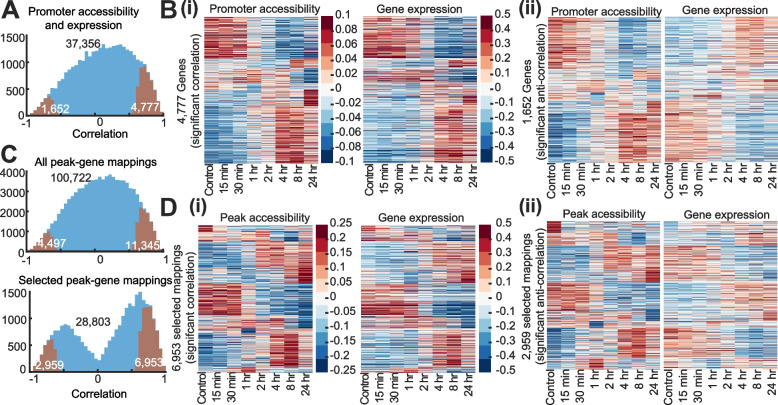
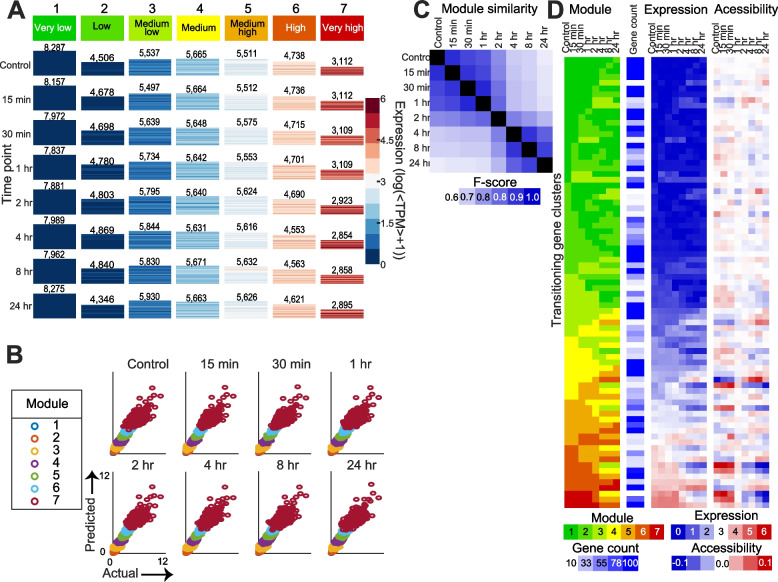
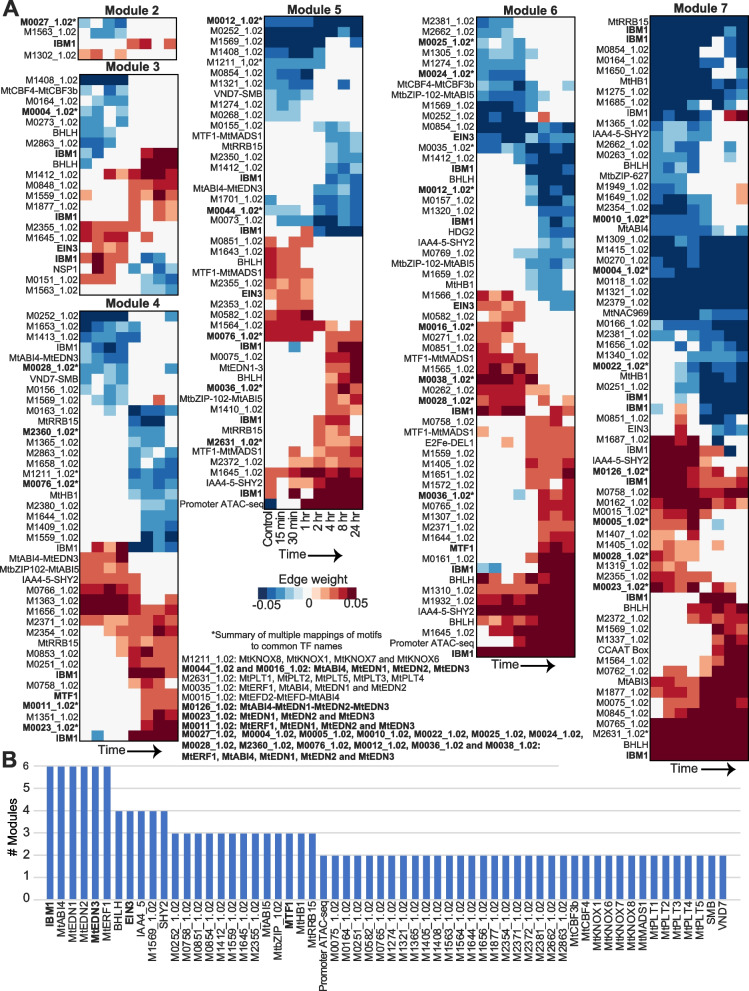
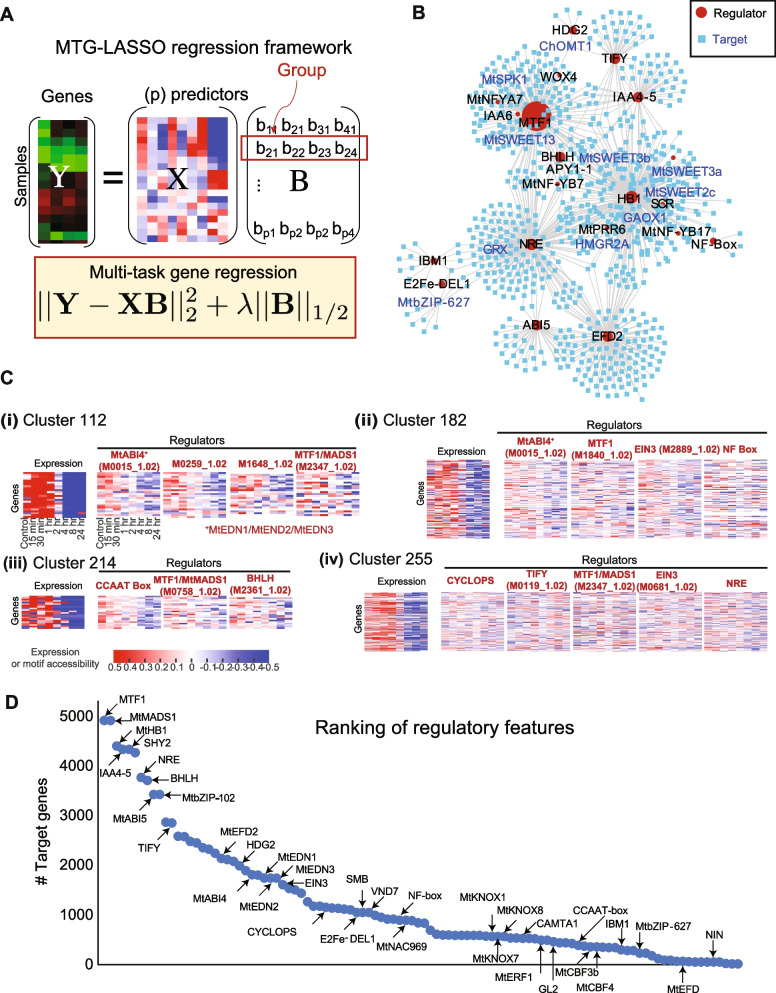
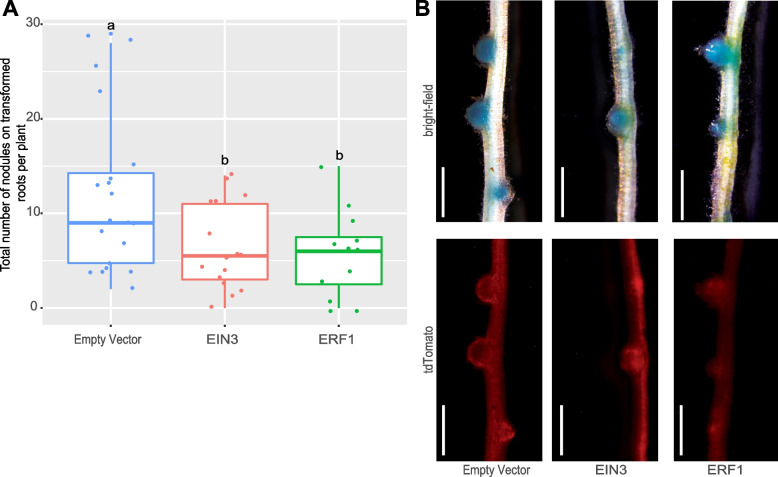
Results: We profiled the temporal chromatin accessibility (ATAC-seq) and transcriptome (RNA-seq) dynamics of M. truncatula roots treated with bacterial small molecules called lipo-chitooligosaccharides that trigger host symbiotic pathways of nodule development. Using a novel approach, dynamic regulatory module networks, we integrated ATAC-seq and RNA-seq time courses to predict cis-regulatory elements and transcription factors that most significantly contribute to transcriptomic changes associated with symbiosis. Regulators involved in auxin (IAA4-5, SHY2), ethylene (EIN3, ERF1), and abscisic acid (ABI5) hormone response, as well as histone and DNA methylation (IBM1), emerged among those most predictive of transcriptome dynamics. RNAi-based knockdown of EIN3 and ERF1 reduced nodule number in M. truncatula validating the role of these predicted regulators in symbiosis between legumes and rhizobia.
Conclusions: Our transcriptomic and chromatin accessibility datasets provide a valuable resource to understand the gene regulatory programs controlling the early stages of the dynamic process of symbiosis. The regulators identified provide potential targets for future experimental validation, and the engineering of nodulation in species is unable to establish that symbiosis naturally.
Keywords: Chromatin accessibility; Cis-regulatory elements; Gene regulatory network; Machine learning; Medicago; Nitrogen fixation; Nodulation; Symbiosis; Transcriptome and chromatin dynamics.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Overexpression of the arginine decarboxylase gene promotes the symbiotic interaction Medicago truncatula-Sinorhizobium meliloti and induces the accumulation of proline and spermine in nodules under salt stress conditions.J Plant Physiol. 2019 Oct;241:153034. doi: 10.1016/j.jplph.2019.153034. Epub 2019 Aug 27. J Plant Physiol. 2019. PMID: 31493718
-
The identification of novel loci required for appropriate nodule development in Medicago truncatula.BMC Plant Biol. 2013 Oct 11;13:157. doi: 10.1186/1471-2229-13-157. BMC Plant Biol. 2013. PMID: 24119289 Free PMC article.
-
Identification of Phytocyanin Gene Family in Legume Plants and their Involvement in Nodulation of Medicago truncatula.Plant Cell Physiol. 2019 Apr 1;60(4):900-915. doi: 10.1093/pcp/pcz007. Plant Cell Physiol. 2019. PMID: 30649463
-
Gene Expression in Nitrogen-Fixing Symbiotic Nodule Cells in Medicago truncatula and Other Nodulating Plants.Plant Cell. 2020 Jan;32(1):42-68. doi: 10.1105/tpc.19.00494. Epub 2019 Nov 11. Plant Cell. 2020. PMID: 31712407 Free PMC article. Review.
-
Leguminous plants: inventors of root nodules to accommodate symbiotic bacteria.Int Rev Cell Mol Biol. 2015;316:111-58. doi: 10.1016/bs.ircmb.2015.01.004. Epub 2015 Feb 20. Int Rev Cell Mol Biol. 2015. PMID: 25805123 Review.
Cited by
-
Autoactive CNGC15 enhances root endosymbiosis in legume and wheat.Nature. 2025 Feb;638(8051):752-759. doi: 10.1038/s41586-024-08424-7. Epub 2025 Jan 15. Nature. 2025. PMID: 39814887 Free PMC article.
-
Functional and comparative genomics reveals conserved noncoding sequences in the nitrogen-fixing clade.New Phytol. 2022 Apr;234(2):634-649. doi: 10.1111/nph.18006. Epub 2022 Feb 21. New Phytol. 2022. PMID: 35092309 Free PMC article.
-
Competence for transcellular infection in the root cortex involves a post-replicative, cell-cycle exit decision in Medicago truncatula.Elife. 2025 Jul 4;12:RP88588. doi: 10.7554/eLife.88588. Elife. 2025. PMID: 40613418 Free PMC article.
References
-
- Smil V. Nitrogen in crop production: an account of global flows. Glob Biogeochem Cycles. 1999;13(2):647–662.
-
- Limpens E, Franken C, Smit P, Willemse J, Bisseling T, Geurts R. LysM domain receptor kinases regulating rhizobial Nod factor-induced infection. Science. 2003;302(5645):630–633. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
