

Genetic analysis of 55 cases with fetal skeletal dysplasia
- PMID: 36352425
- PMCID: PMC9648031
- DOI: 10.1186/s13023-022-02559-4
Genetic analysis of 55 cases with fetal skeletal dysplasia
Abstract
Background: Fetal skeletal dysplasia (SD) is a common congenital disability comprising a complex group of skeletal disorders with substantial clinical and genetic heterogeneity. Many of these defects are detected prenatally using ultrasound (US). However, the diagnostic accuracy of the US is limited.
Methods: We recruited 55 unrelated fetuses with US-detected skeletal anomalies and performed sequential tests using copy number variation sequencing, targeted skeletal gene panel sequencing, or whole exome sequencing. The detected variants were validated using Sanger sequencing or multiplex ligation-dependent probe amplification. We conducted breakpoint analysis and structural modeling of variants possibly involved in fetal SD.
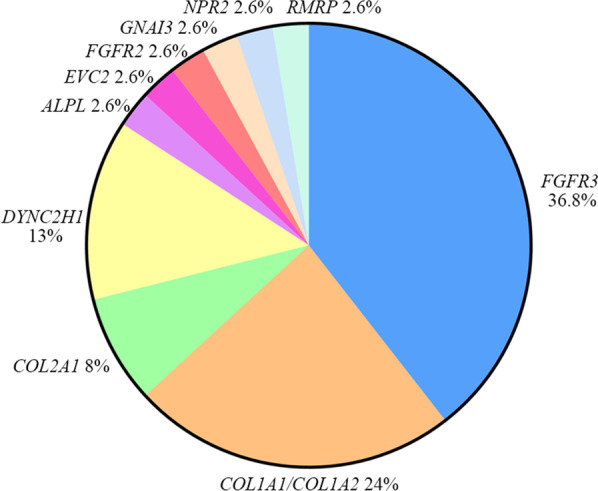
Results: A definitive diagnosis was achieved in 81.82% of affected fetuses (45/55). We identified chromosomal abnormalities in seven cases and 36 variants, of which 18 were novel pathogenic or likely pathogenic in 11 genes in 38 cases. De novo variants were identified in 27 cases (71.05%, 27/38), and one gonosomal mosaicism variant was found in the mother of one fetus. Our case examples demonstrated the high heterogeneity of fetal SDs and the rare fetal SD-associated challenges.
Conclusions: Careful clinical evaluation of fetuses with SD can guide appropriate molecular testing. Our study extends the SD-associated pathogenic variant spectrum and provides useful genetic counselling guidance and an accurate prenatal diagnosis strategy.
Keywords: Breakpoints; Genotype–phenotype analysis; Molecular diagnosis; Novel variants; Skeletal dysplasia.
© 2022. The Author(s).
Conflict of interest statement
The authors have declared no conflicting interests.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
