

Personalized genome assembly for accurate cancer somatic mutation discovery using tumor-normal paired reference samples
- PMID: 36352452
- PMCID: PMC9648002
- DOI: 10.1186/s13059-022-02803-x
Personalized genome assembly for accurate cancer somatic mutation discovery using tumor-normal paired reference samples
Abstract
Background: The use of a personalized haplotype-specific genome assembly, rather than an unrelated, mosaic genome like GRCh38, as a reference for detecting the full spectrum of somatic events from cancers has long been advocated but has never been explored in tumor-normal paired samples. Here, we provide the first demonstrated use of de novo assembled personalized genome as a reference for cancer mutation detection and quantifying the effects of the reference genomes on the accuracy of somatic mutation detection.
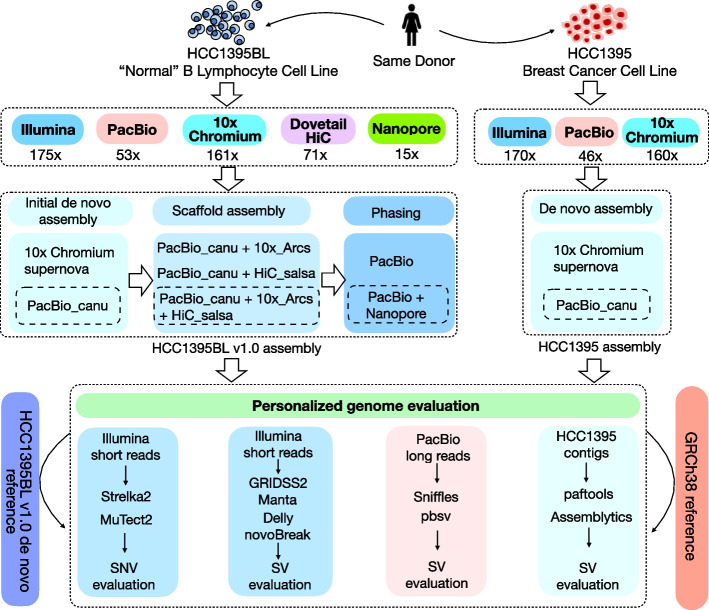
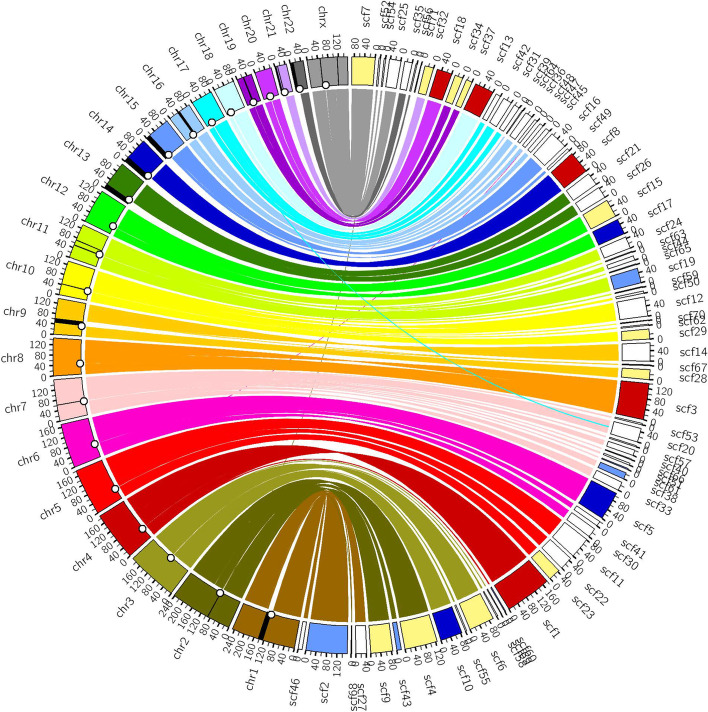
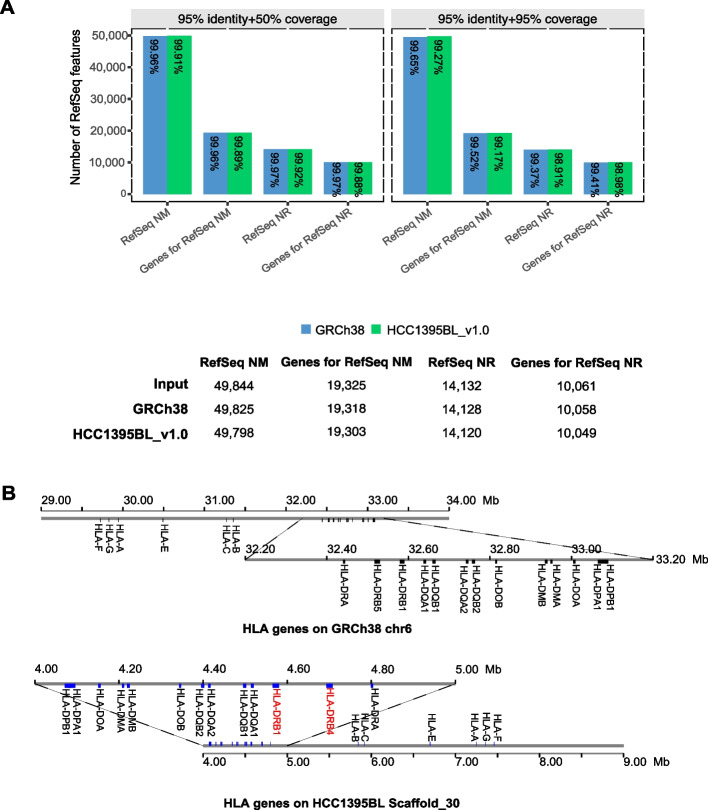
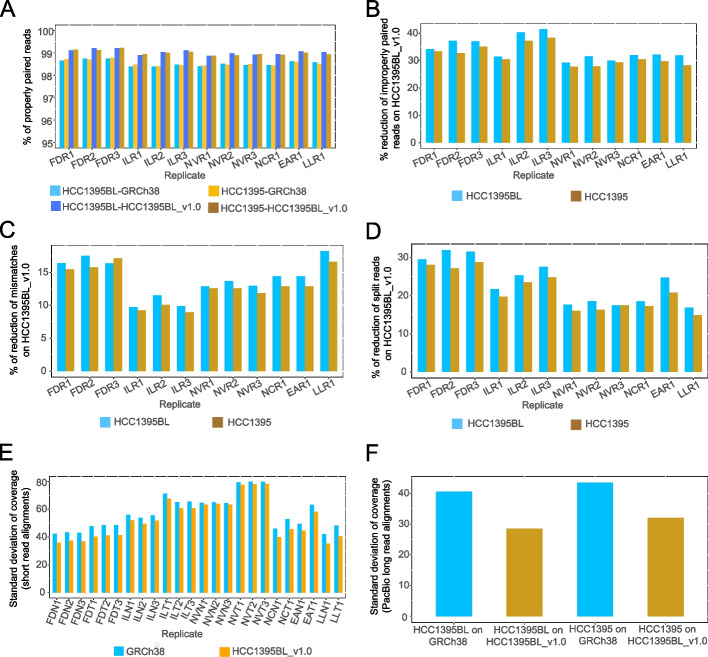
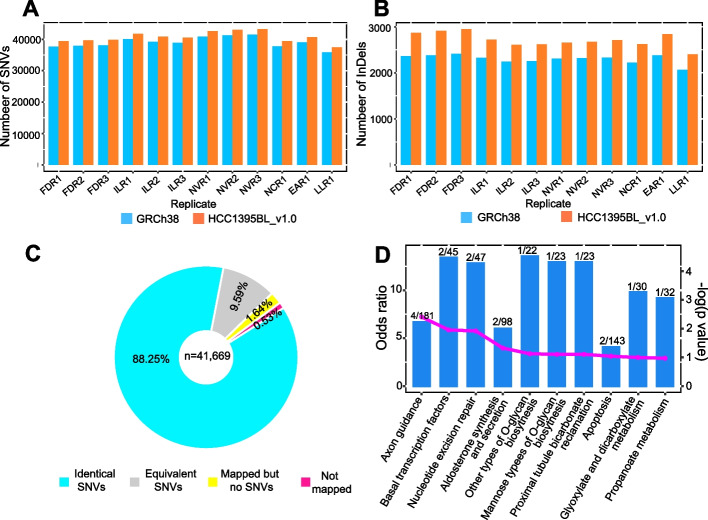
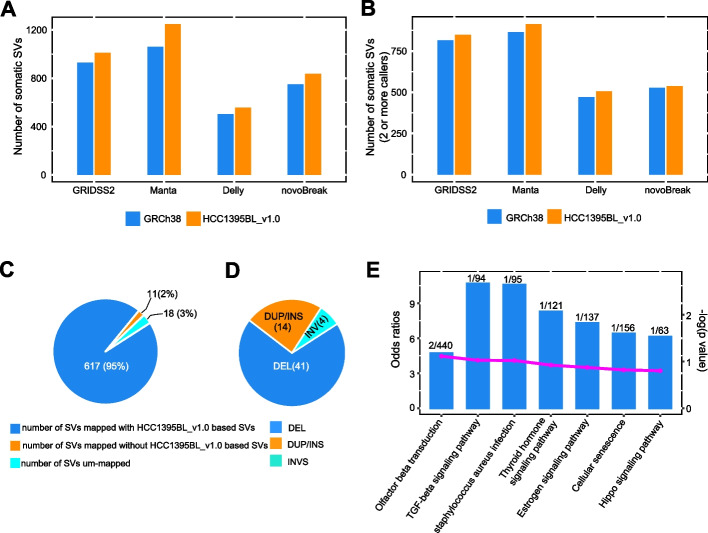
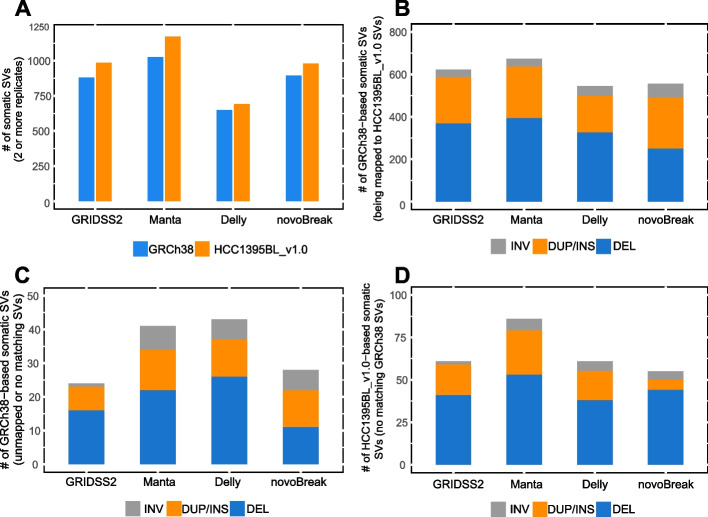
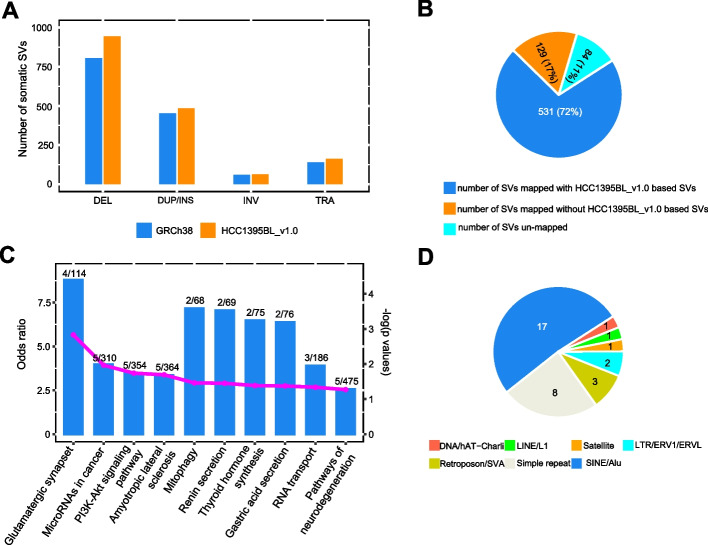
Results: We generate de novo assemblies of the first tumor-normal paired genomes, both nuclear and mitochondrial, derived from the same individual with triple negative breast cancer. The personalized genome was chromosomal scale, haplotype phased, and annotated. We demonstrate that it provides individual specific haplotypes for complex regions and medically relevant genes. We illustrate that the personalized genome reference not only improves read alignments for both short-read and long-read sequencing data but also ameliorates the detection accuracy of somatic SNVs and SVs. We identify the equivalent somatic mutation calls between two genome references and uncover novel somatic mutations only when personalized genome assembly is used as a reference.
Conclusions: Our findings demonstrate that use of a personalized genome with individual-specific haplotypes is essential for accurate detection of the full spectrum of somatic mutations in the paired tumor-normal samples. The unique resource and methodology established in this study will be beneficial to the development of precision oncology medicine not only for breast cancer, but also for other cancers.
© 2022. The Author(s).
Conflict of interest statement
CP was employed by Dovetail Genomics, LLC, and LF was employed by Roche Sequencing Solutions Inc during the course of this research. The other authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
