

Natural Populations from the Phytophthora palustris Complex Show a High Diversity and Abundance of ssRNA and dsRNA Viruses
- PMID: 36354885
- PMCID: PMC9698713
- DOI: 10.3390/jof8111118
Natural Populations from the Phytophthora palustris Complex Show a High Diversity and Abundance of ssRNA and dsRNA Viruses
Abstract
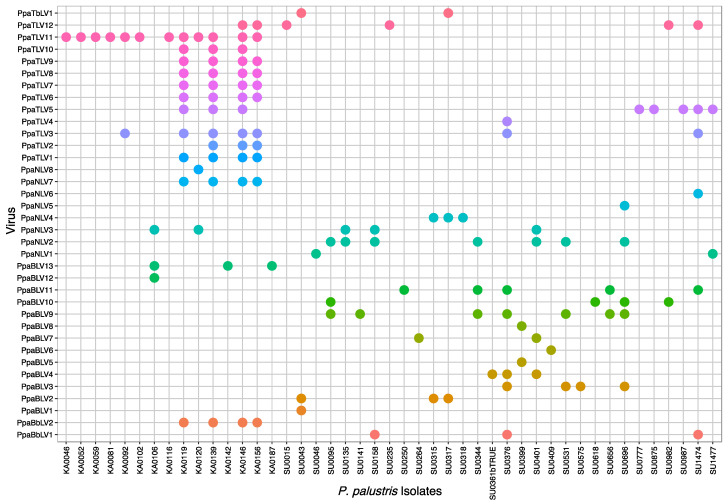
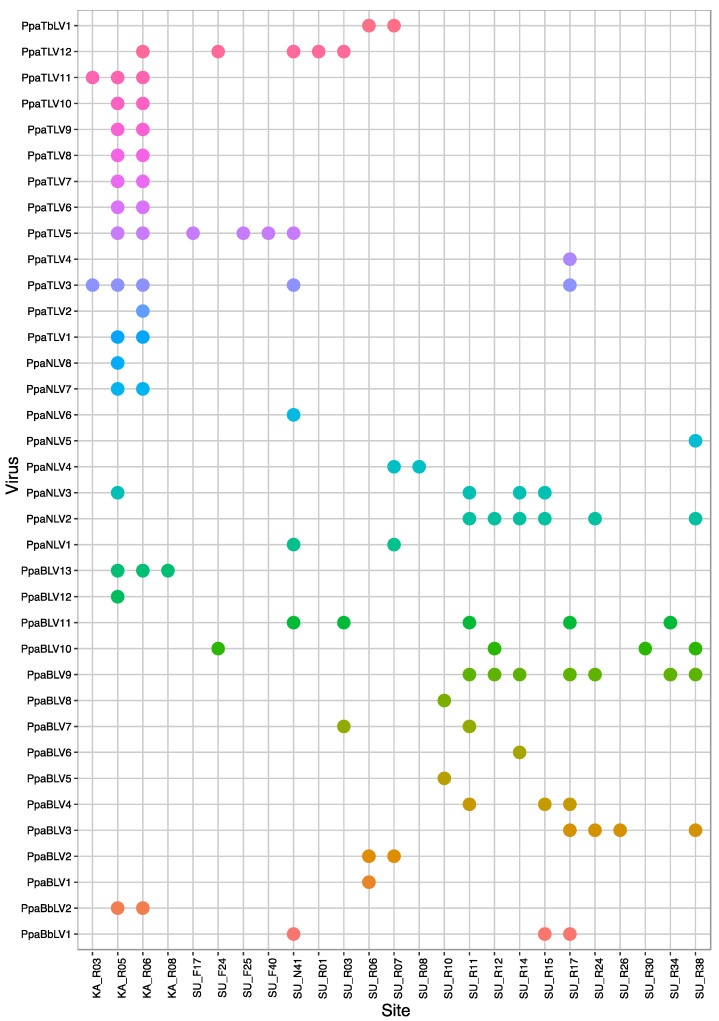
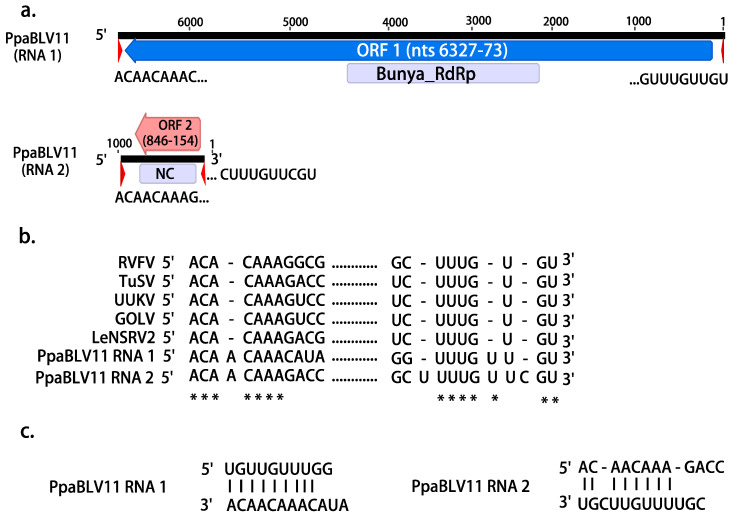
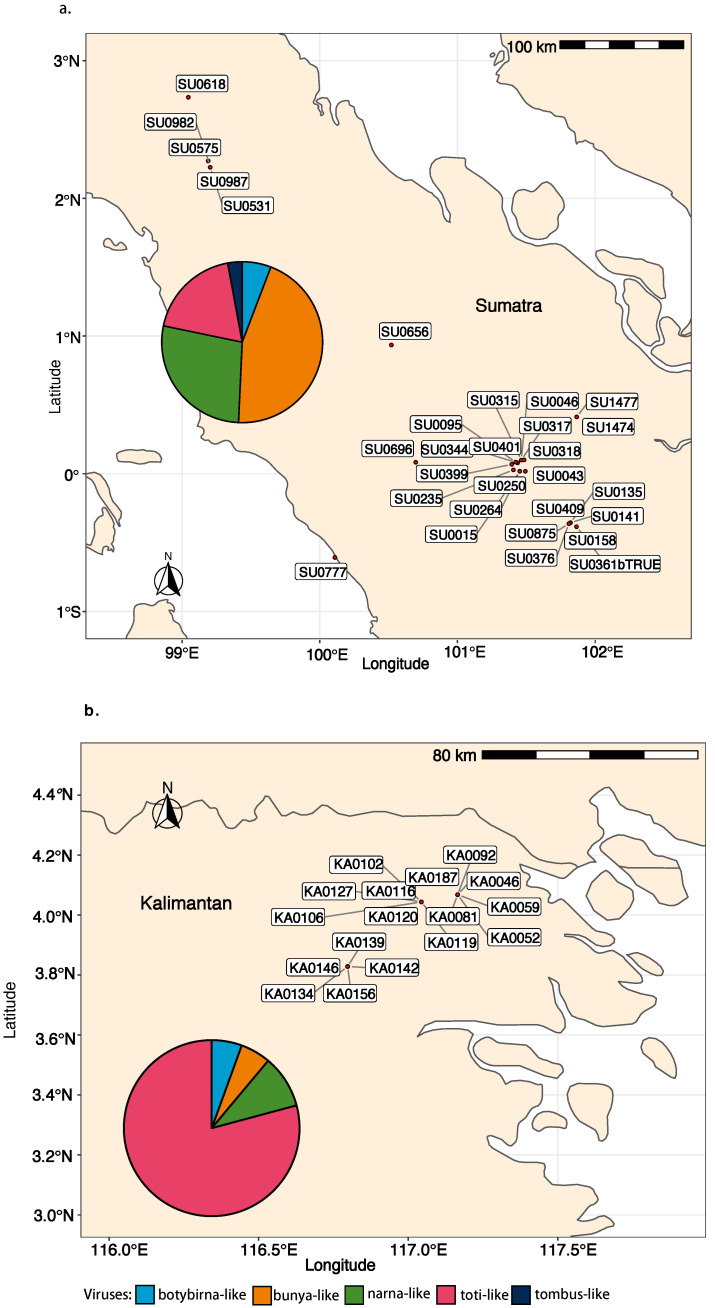
We explored the virome of the "Phytophthora palustris complex", a group of aquatic specialists geographically limited to Southeast and East Asia, the native origin of many destructive invasive forest Phytophthora spp. Based on high-throughput sequencing (RNAseq) of 112 isolates of "P. palustris" collected from rivers, mangroves, and ponds, and natural forests in subtropical and tropical areas in Indonesia, Taiwan, and Japan, 52 putative viruses were identified, which, to varying degrees, were phylogenetically related to the families Botybirnaviridae, Narnaviridae, Tombusviridae, and Totiviridae, and the order Bunyavirales. The prevalence of all viruses in their hosts was investigated and confirmed by RT-PCR. The rich virus composition, high abundance, and distribution discovered in our study indicate that viruses are naturally infecting taxa from the "P. palustris complex" in their natural niche, and that they are predominant members of the host cellular environment. Certain Indonesian localities are the viruses' hotspots and particular "P. palustris" isolates show complex multiviral infections. This study defines the first bi-segmented bunya-like virus together with the first tombus-like and botybirna-like viruses in the genus Phytophthora and provides insights into the spread and evolution of RNA viruses in the natural populations of an oomycete species.
Keywords: Phytophthora; RNA-sequencing; multiple viral infections; mycovirus; natural habitat; oomycetes; virus ecology; virus evolution; virus reservoirs.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
 Fungi,
Fungi,  Nematoda,
Nematoda,  Oomycota,
Oomycota,  Arthropoda,
Arthropoda,  Plants,
Plants,  Mammalia,
Mammalia,  (Fishes) Chordata,
(Fishes) Chordata,  Ochrophyta (Heterokonta),
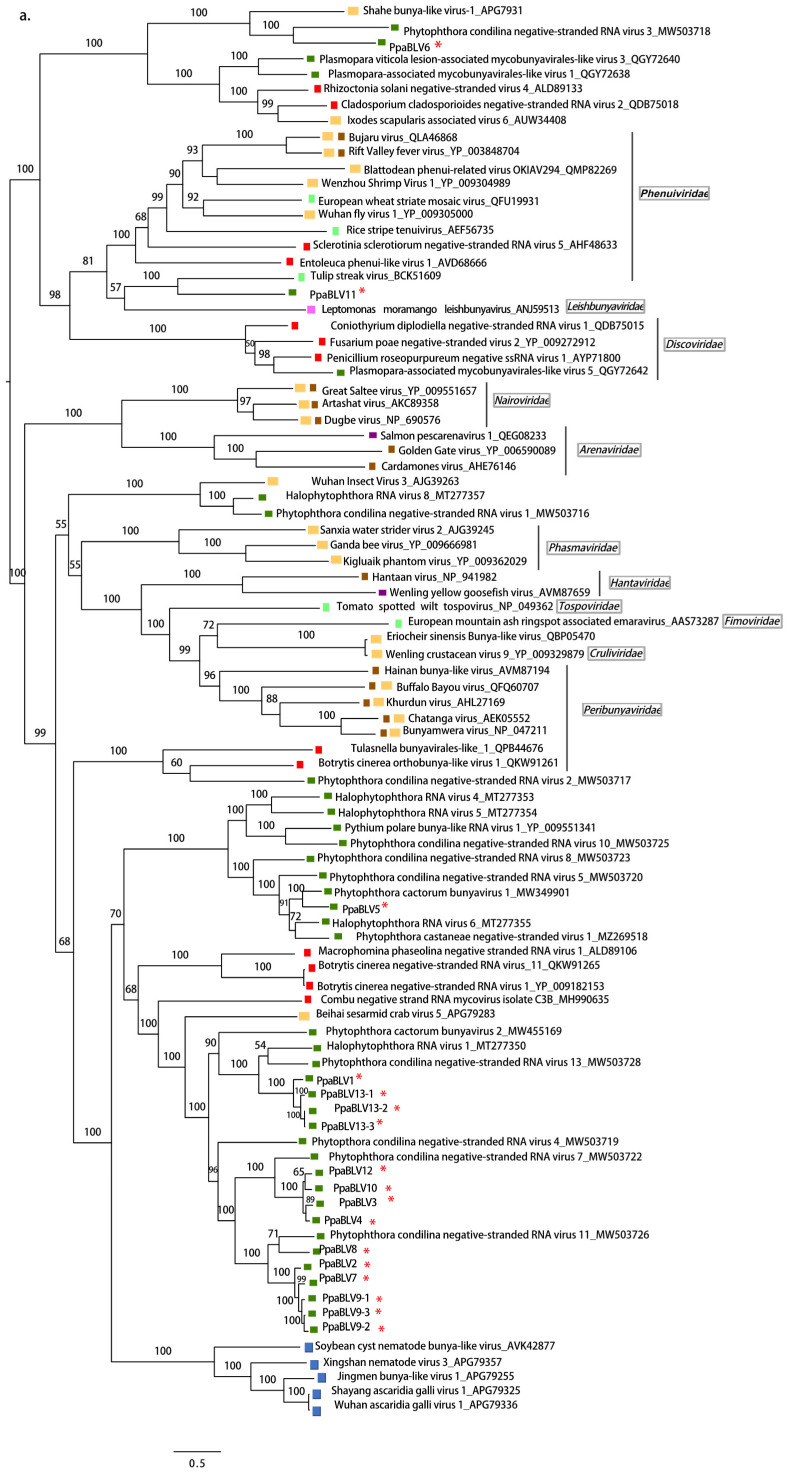
Ochrophyta (Heterokonta),  Excavata. Scale bars represent expected changes per site per branch.
Excavata. Scale bars represent expected changes per site per branch.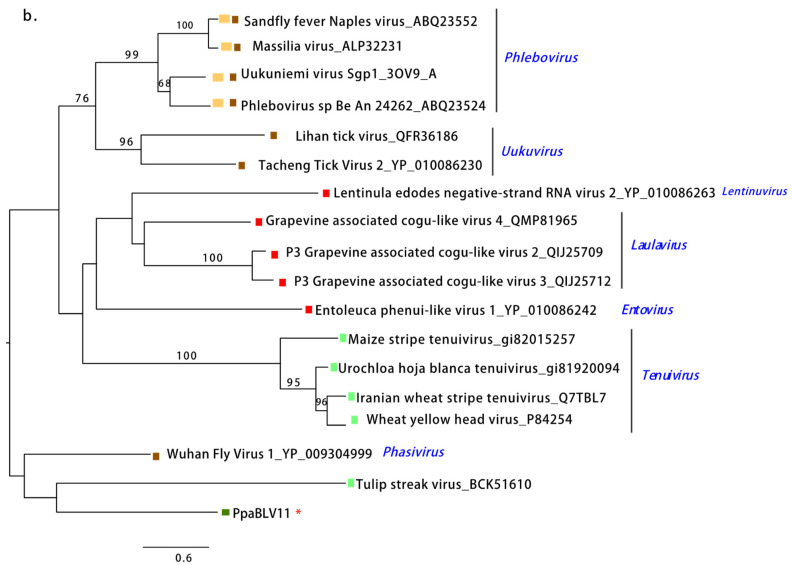 Fungi, Nematoda, Oomycota, Arthropoda, Plants, Mammalia, (Fishes) Chordata, Ochrophyta (Heterokonta), Excavata. Scale bars represent expected changes per site per branch.
Fungi, Nematoda, Oomycota, Arthropoda, Plants, Mammalia, (Fishes) Chordata, Ochrophyta (Heterokonta), Excavata. Scale bars represent expected changes per site per branch.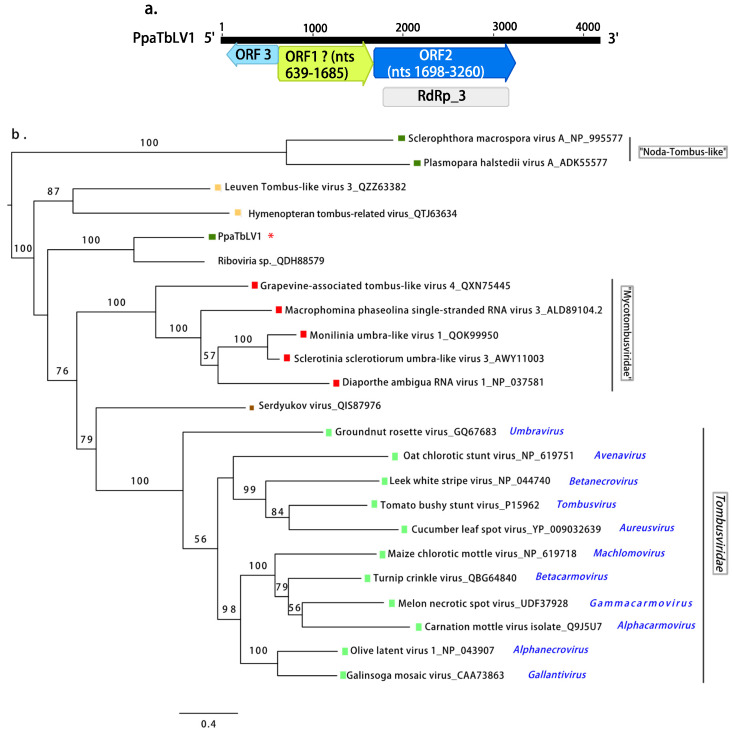 Fungi, Oomycota, Arthropoda,
Fungi, Oomycota, Arthropoda,  Animalia, Plants. Scale bar = 0.4 expected changes per site per branch.
Animalia, Plants. Scale bar = 0.4 expected changes per site per branch.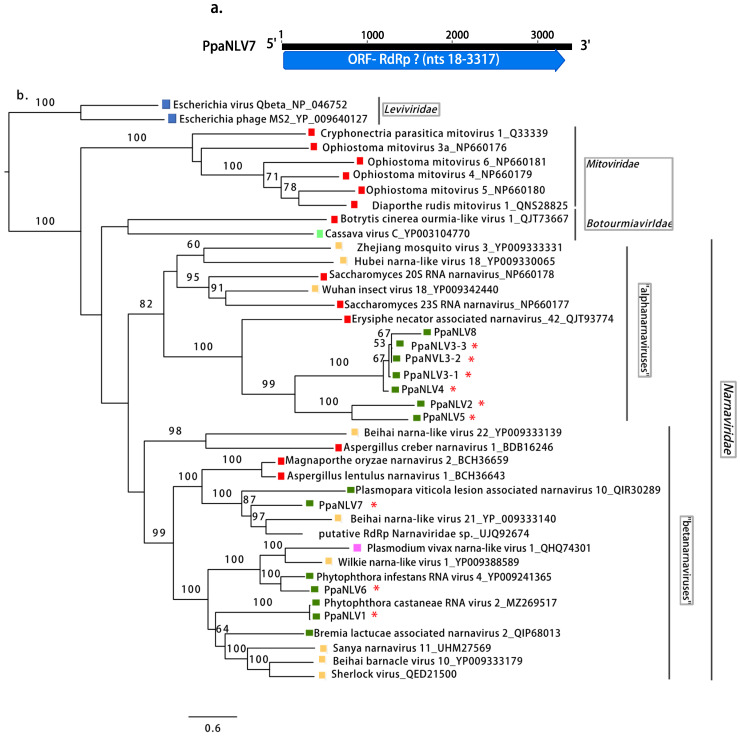 Fungi, Oomycota, Arthropoda, Animalia, Plants, Ochrophyta (Heterokonta),
Fungi, Oomycota, Arthropoda, Animalia, Plants, Ochrophyta (Heterokonta),  Apicomplexa (Protists),
Apicomplexa (Protists),  Bacteria. Scale bar = 0.6 expected changes per site per branch.
Bacteria. Scale bar = 0.6 expected changes per site per branch. Fungi, Oomycota, Arthropoda, Chordata, Ochrophyta (Heterokonta), Excavata. Scale bar = 0.6 expected changes per site per branch.
Fungi, Oomycota, Arthropoda, Chordata, Ochrophyta (Heterokonta), Excavata. Scale bar = 0.6 expected changes per site per branch.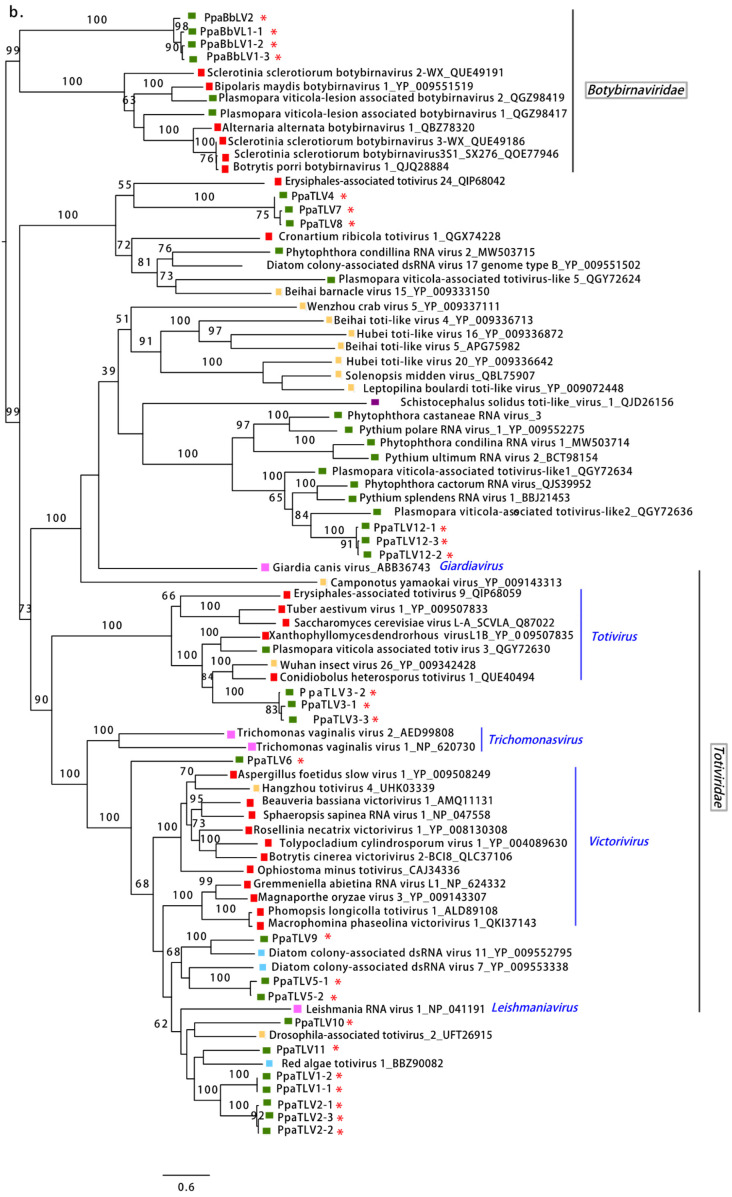 Fungi, Oomycota, Arthropoda, Chordata, Ochrophyta (Heterokonta), Excavata. Scale bar = 0.6 expected changes per site per branch.
Fungi, Oomycota, Arthropoda, Chordata, Ochrophyta (Heterokonta), Excavata. Scale bar = 0.6 expected changes per site per branch.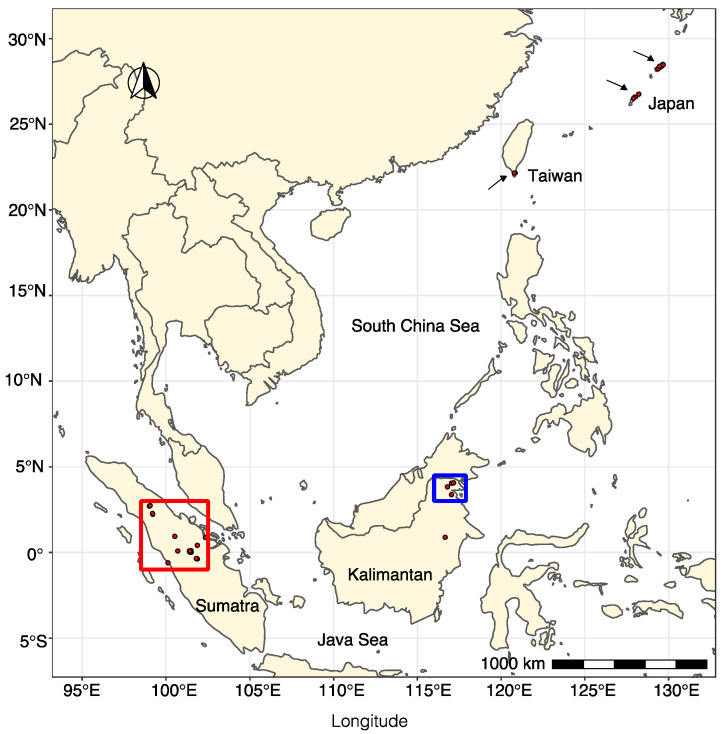
References
-
- Brasier C.M. The biosecurity threat to the UK and global environment from international trade in plants. Plant Pathol. 2008;57:792–808. doi: 10.1111/j.1365-3059.2008.01886.x. - DOI
-
- Santini A., Ghelardini L., De Pace C., Desprez-Loustau M.L., Capretti P., Chandelier A., Cech T., Chira D., Diamandis S., Gaitniekis T., et al. Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytol. 2013;197:238–250. doi: 10.1111/j.1469-8137.2012.04364.x. - DOI - PubMed
-
- Hantula J., Müller M.M., Uusivuori J. International plant trade associated risks: Laissez-faire or novel solutions. Environ. Sci. Policy. 2014;37:158–160. doi: 10.1016/j.envsci.2013.09.011. - DOI
-
- Jung T., Orlikowski L., Henricot B., Abad-Campos P., Aday A.G., Aguín Casal O., Bakonyi J., Cacciola S.O., Cech T., Chavarriaga D., et al. Widespread Phytophthora infestations in European nurseries put forest, semi-natural and horticultural ecosystems at high risk of Phytophthora diseases. For. Pathol. 2016;46:134–163. doi: 10.1111/efp.12239. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
