

Dysregulated Lipid Synthesis by Oncogenic IDH1 Mutation Is a Targetable Synthetic Lethal Vulnerability
- PMID: 36355448
- PMCID: PMC9900324
- DOI: 10.1158/2159-8290.CD-21-0218
Dysregulated Lipid Synthesis by Oncogenic IDH1 Mutation Is a Targetable Synthetic Lethal Vulnerability
Abstract
Isocitrate dehydrogenase 1 and 2 (IDH) are mutated in multiple cancers and drive production of (R)-2-hydroxyglutarate (2HG). We identified a lipid synthesis enzyme [acetyl CoA carboxylase 1 (ACC1)] as a synthetic lethal target in mutant IDH1 (mIDH1), but not mIDH2, cancers. Here, we analyzed the metabolome of primary acute myeloid leukemia (AML) blasts and identified an mIDH1-specific reduction in fatty acids. mIDH1 also induced a switch to b-oxidation indicating reprogramming of metabolism toward a reliance on fatty acids. Compared with mIDH2, mIDH1 AML displayed depletion of NADPH with defective reductive carboxylation that was not rescued by the mIDH1-specific inhibitor ivosidenib. In xenograft models, a lipid-free diet markedly slowed the growth of mIDH1 AML, but not healthy CD34+ hematopoietic stem/progenitor cells or mIDH2 AML. Genetic and pharmacologic targeting of ACC1 resulted in the growth inhibition of mIDH1 cancers not reversible by ivosidenib. Critically, the pharmacologic targeting of ACC1 improved the sensitivity of mIDH1 AML to venetoclax.
Significance: Oncogenic mutations in both IDH1 and IDH2 produce 2-hydroxyglutarate and are generally considered equivalent in terms of pathogenesis and targeting. Using comprehensive metabolomic analysis, we demonstrate unexpected metabolic differences in fatty acid metabolism between mutant IDH1 and IDH2 in patient samples with targetable metabolic interventions. See related commentary by Robinson and Levine, p. 266. This article is highlighted in the In This Issue feature, p. 247.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
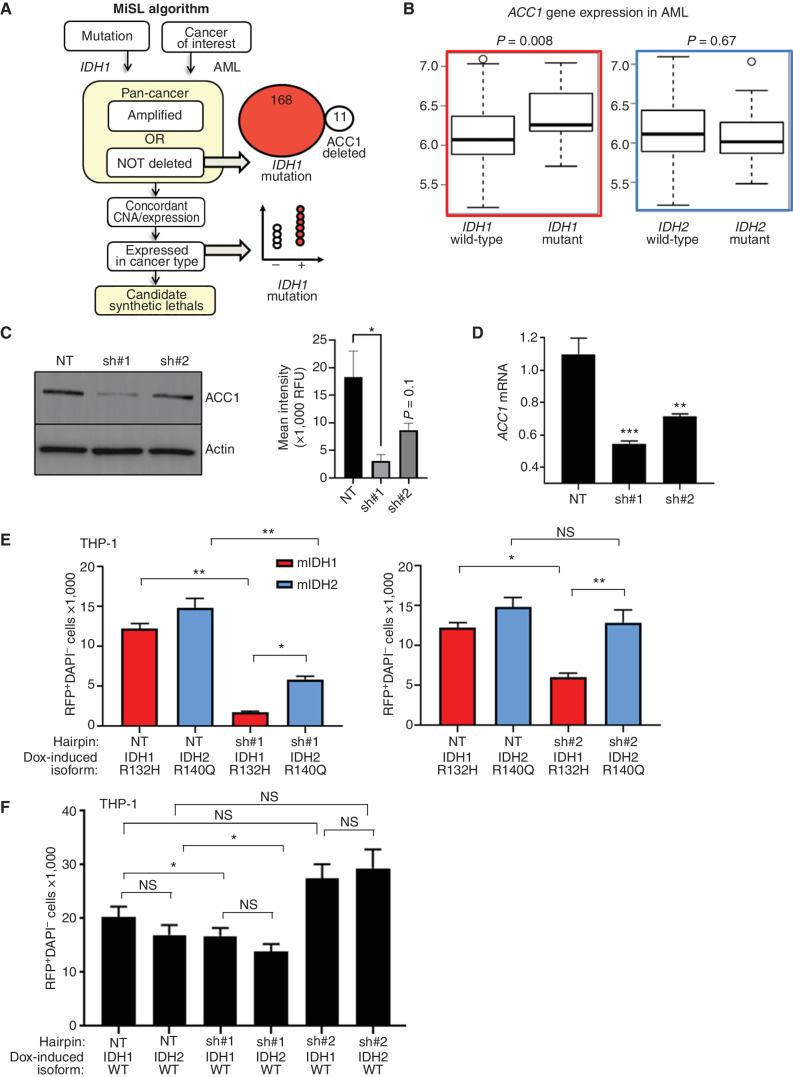
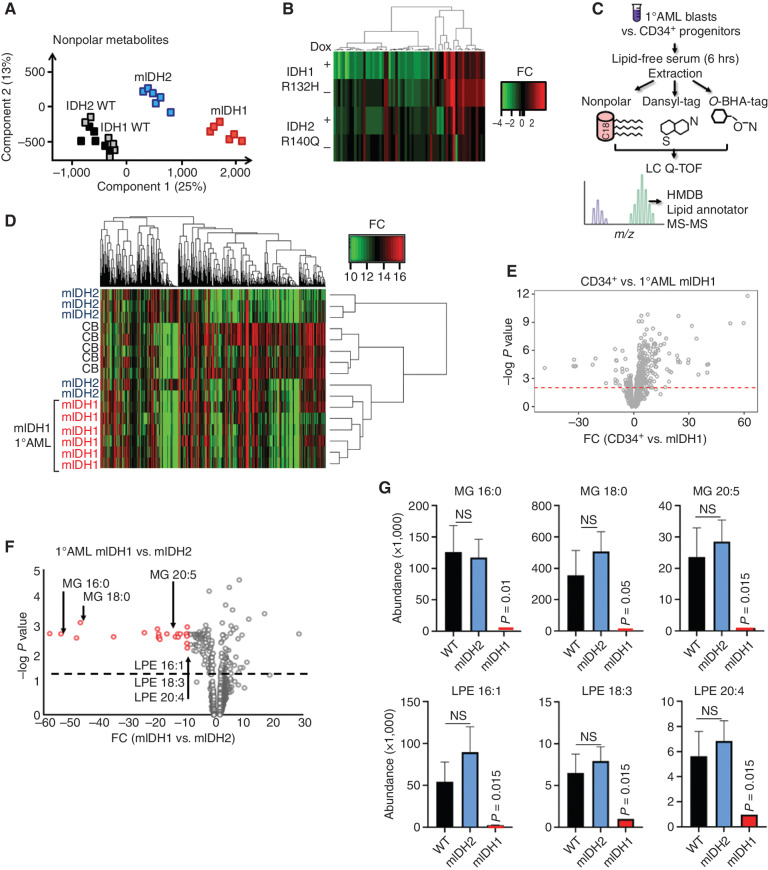
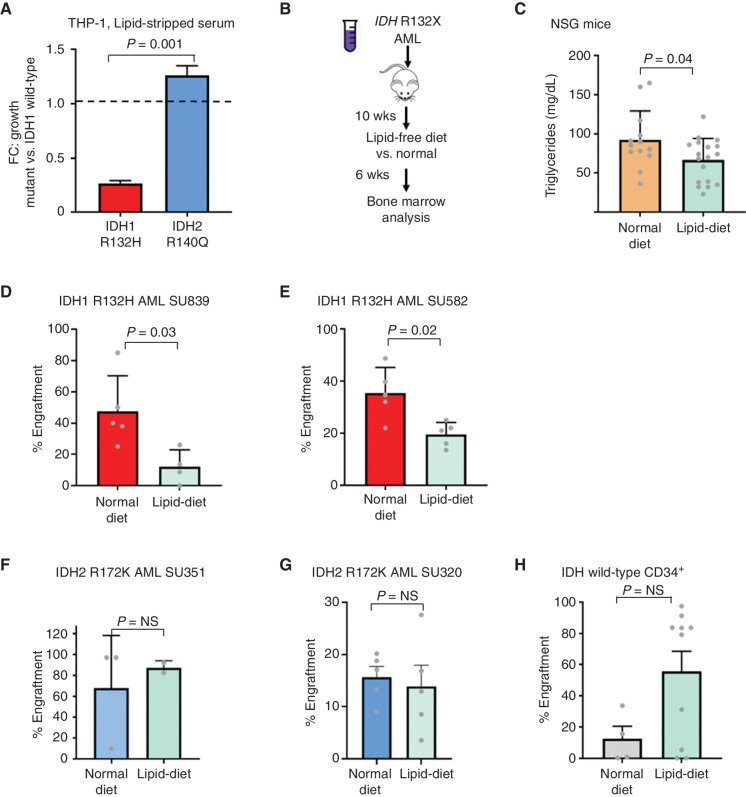
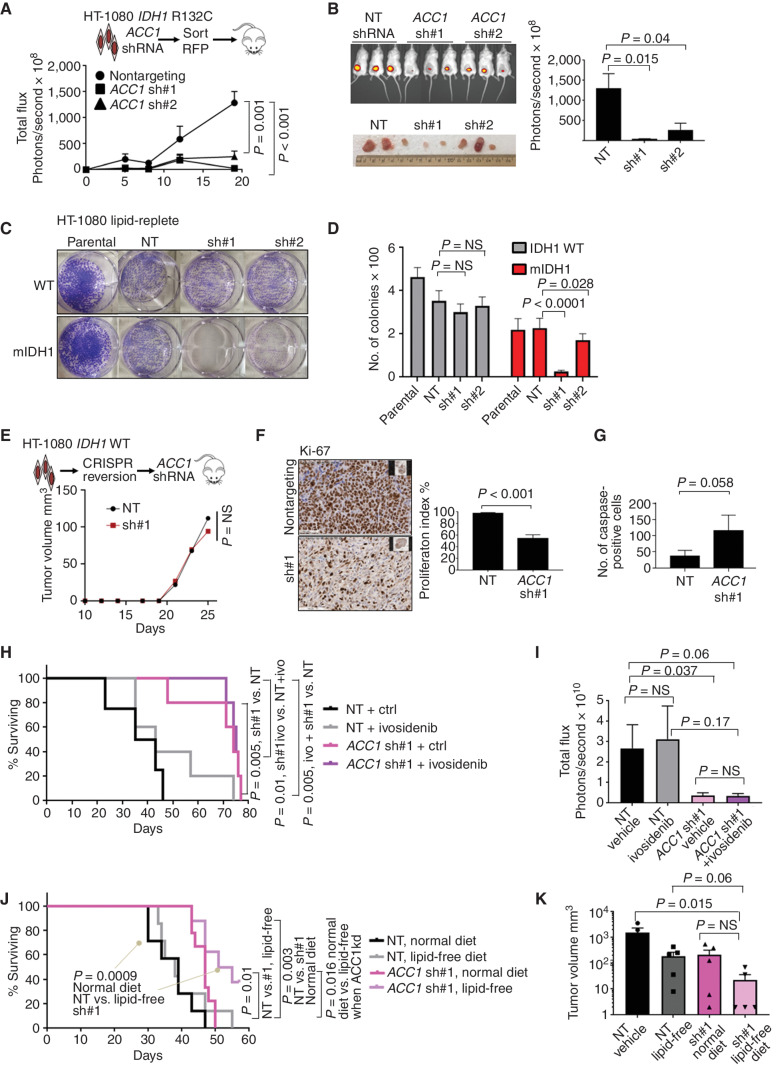
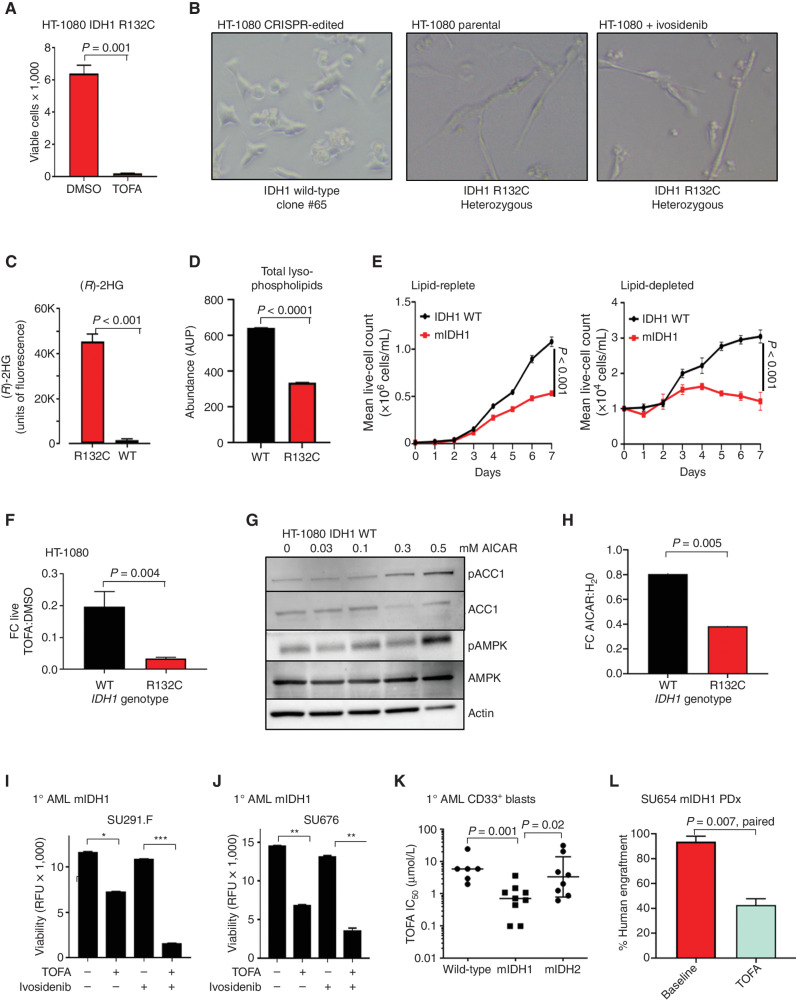
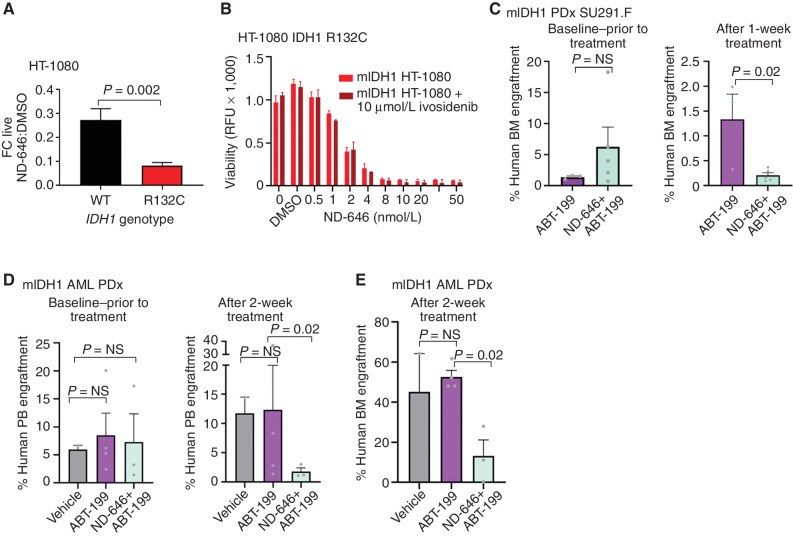
![Figure 3. IDH1 mutation is linked to defective reductive carboxylation, increased fatty acid consumption, and decreased NADPH. A, Schematic and graph of flux studies showing the percentage of labeled M2 glycerol-3-phosphate derived from 13C[1,2] labeled glucose (2 of 6 carbons as heavy isotope) across THP-1 cells induced to express mIDH1 vs. IDH1 wild-type compared with mIDH2 vs. IDH2 wild-type. Glucose was added to media in normoxia over 13 hours. Schematic indicates M2 isotopolog of glycerol-3-phosphate de novo synthesis directly from glycolysis utilizing labeled glucose rather than the oxidative pentose phosphate pathway (M1 isotopolog). A two-tailed unpaired Student t test was used to compare differences between groups. This experiment was performed with 6 cell pellets for each sample blinded and randomized on each LC-MS run. B, Decreased reductive carboxylation of mIDH1 compared with IDH1 wild-type and mIDH2 expressed in THP-1 cells measured by the percentage of M5 citrate isotopolog obtained from U-13C5 glutamine labeling in 2% hypoxia over 16 hours as shown in the schematic. The last bar shows mIDH1 cells cultured in the presence of 10 μmol/L ivosidenib added prior to adding a label. This experiment was performed with 6 cell pellets for each sample blinded and randomized on each LC-MS run; ***, P < 0.001; **, P < 0.01, Student t test. C, Column graph showing fold change increase in acylcarnitine metabolites after induction of mIDH1 (+dox) vs. wild-type (−dox) in comparison with mIDH2 (+dox) vs. IDH2 wild-type (−dox) in THP-1 cells as measured by LC-MS. Student t test is used to compare groups. D, Column graph showing mean fatty acid β-oxidation as measured by the etomoxir-sensitive component of oxygen consumption in pmole/min/105 cells in mIDH1 vs. IDH1 wild-type, mIDH2 and IDH2 wild-type in THP-1 cells measured on Seahorse analyzer, 3 independent experiments. E, Column graph showing the percentage of beta-oxidation of total oxygen consumption (etomoxir-sensitive component) in mIDH1 and mIDH2 before and after the addition of the ACC1 inhibitor 100 nmol/L ND-646. A representative experiment is shown. Statistics represent paired t test with n = 4 replicates. F, Column graph showing % etomoxir-sensitive component in mIDH1 before and after the addition of 10 μmol/L ivosidenib. P = nonsignificant, Student t test, 4 replicates. G, Western blot showing phospho-AMPK on threonine 172 in THP-1 cells induced with mIDH1, mIDH2 and wild-type counterparts in lipid-replete conditions. Comparison with total AMPK alpha isoform and actin is shown in panels below. H, NADPH levels measured in identical 2 × 106 viable cell pellets of mIDH1, mIDH2, or wild-type primary AML blasts vs. normal CD34+ cells grown in culture for 48 hours in normoxia. Symbols indicate individual sample values. Student t test is used to compare groups. I, Schematic summarizing mIDH1-induced mechanisms impacting lipid metabolism. Pathways involved directly in phospholipid synthesis that are perturbed by mIDH1 are shown in red. Thin arrows indicate reduced flux, and thick arrows indicate preserved or increased flux. Black arrows indicate pathways not measurably affected by mIDH1. Mechanistic causes for aberrant lipid metabolism identified in our study include: (i) reduced carbon flux arising from defective reductive carboxylation by mutant IDH1 hetero/homodimers exacerbated in relative marrow hypoxia and mitochondrial stress, (ii) cumulative NADP(H) decrease by neomorphic reverse αKG to 2HG reaction, (iii) ongoing depletion of NADPH by residual reductive carboxylation of glutamine, (iv) insufficient NADPH replenishment by impaired forward reaction akin to TCA cycle, (v) increased fatty acid β-oxidation with a concomitant increase in acylcarnitines that is sensitive to ACC1 inhibition but not ivosidenib, and (vi) decreased AMPK phosphorylation, indicating an AMPK-independent mechanism for enhanced oxidative phosphorylation of fatty acids. In contrast, the flux of glucose to produce glycerol-3-phosphate, the building block for the glycerol component of glycerolipids, is not impaired and is rather upregulated in mIDH1 compared with mIDH2, shown in black. This would suggest de novo synthesis of glycerol from glucose is not the major cause of defective lipid species in mIDH1 AML. Similarly, static levels of citric acid cycle metabolites are decreased to similar in both mIDH1 and mIDH2 AML, as shown in Supplementary figures. Blockade of 2HG production by ivosidenib is not sufficient for restoring defective reductive carboxylation nor abrogating β-oxidation.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/8f73/9900324/036f12733f94/496fig3.jpg)
Comment in
-
Oncogenic IDH1 Mutation Imparts Therapeutically Targetable Metabolic Dysfunction in Multiple Tumor Types.Cancer Discov. 2023 Feb 6;13(2):266-268. doi: 10.1158/2159-8290.CD-22-1325. Cancer Discov. 2023. PMID: 36744320
References
-
- Chou W, Hou H, Chen C, Tang J, Yao M, Tsay W, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 2010;115:2749–54. - PubMed
-
- Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med 2010;16:387–97. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
