

A novel RIPK1 inhibitor reduces GVHD in mice via a nonimmunosuppressive mechanism that restores intestinal homeostasis
- PMID: 36356302
- PMCID: PMC10651787
- DOI: 10.1182/blood.2022017262
A novel RIPK1 inhibitor reduces GVHD in mice via a nonimmunosuppressive mechanism that restores intestinal homeostasis
Abstract
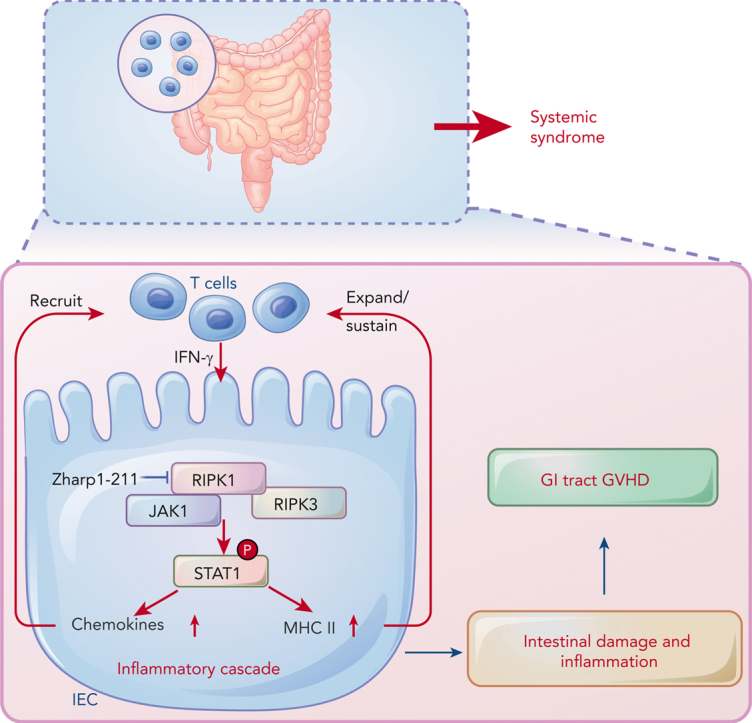
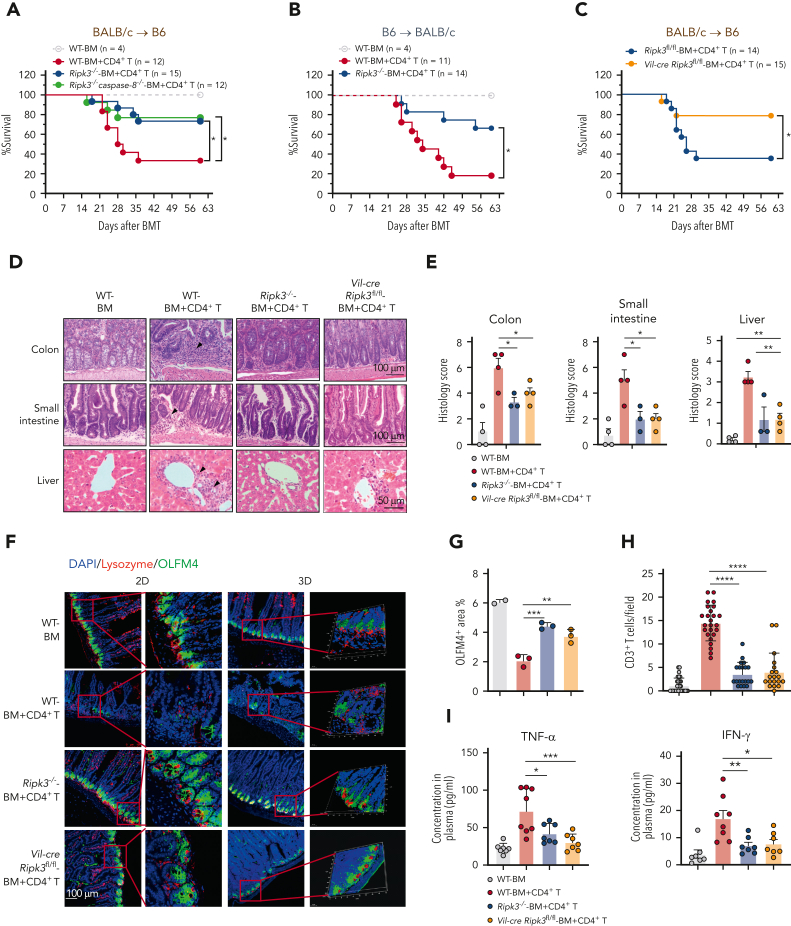
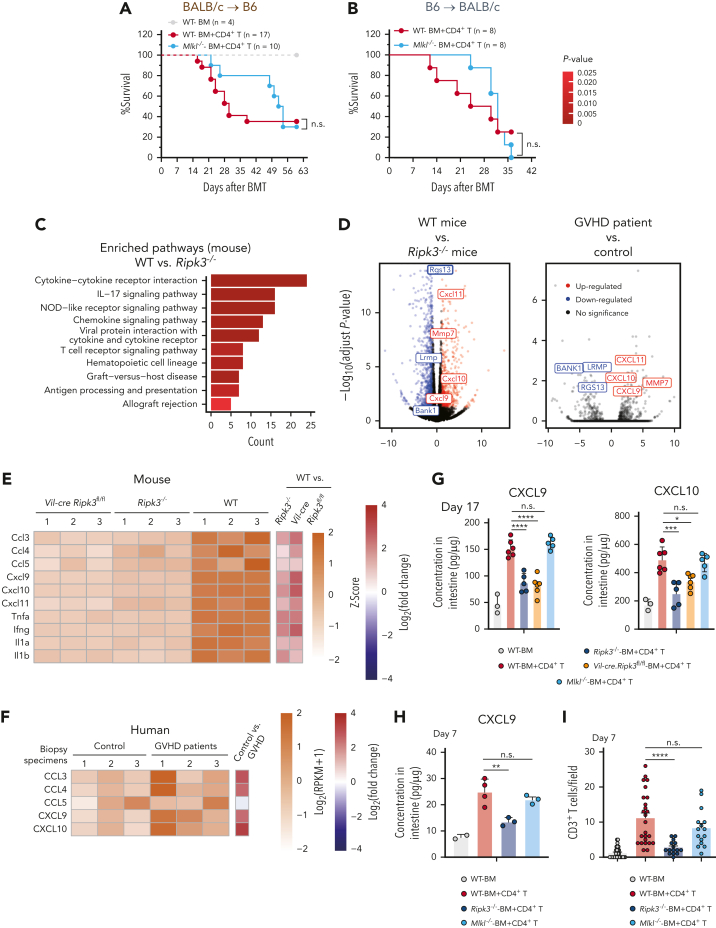
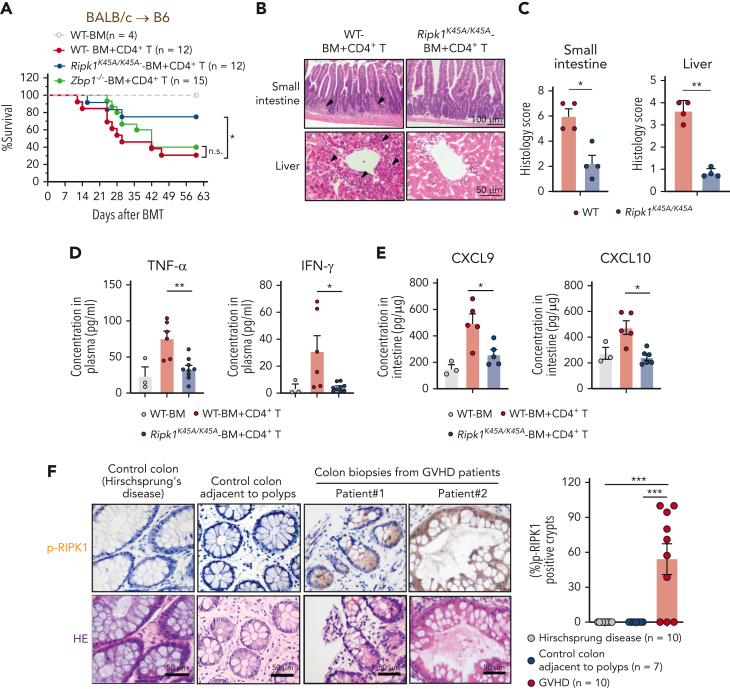
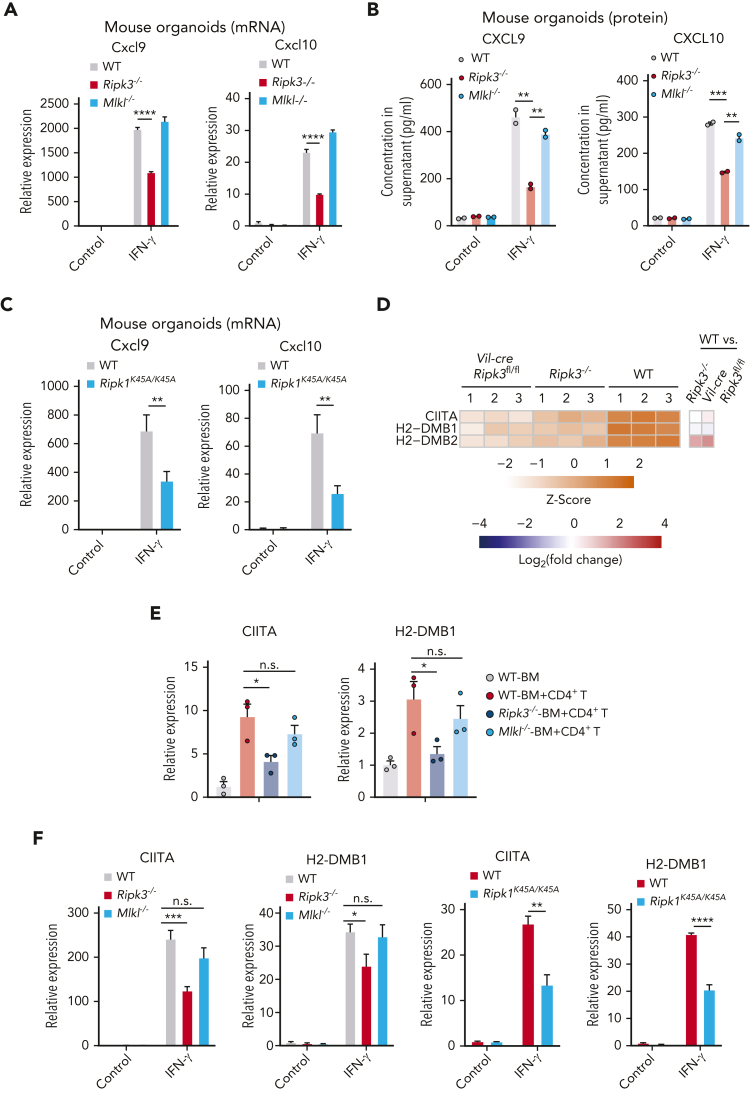
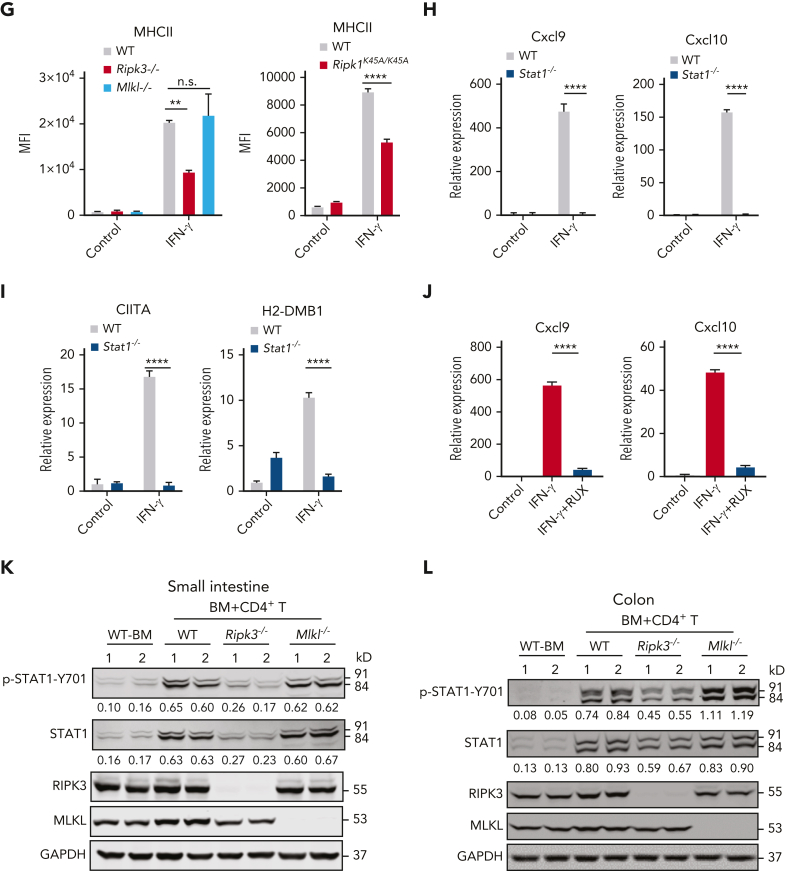
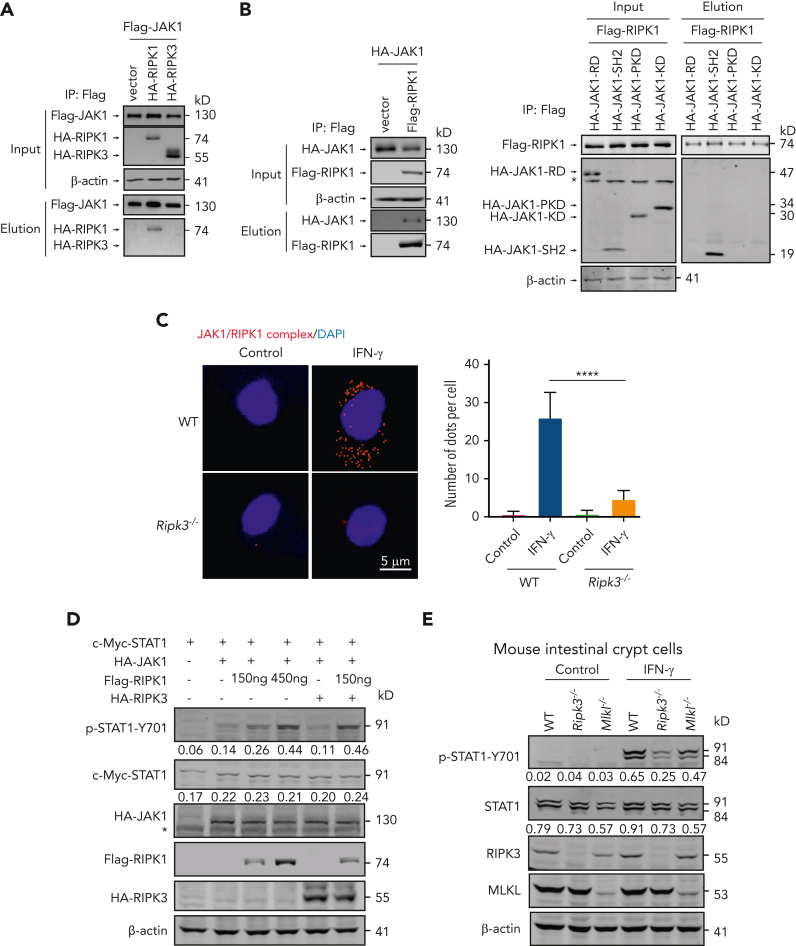
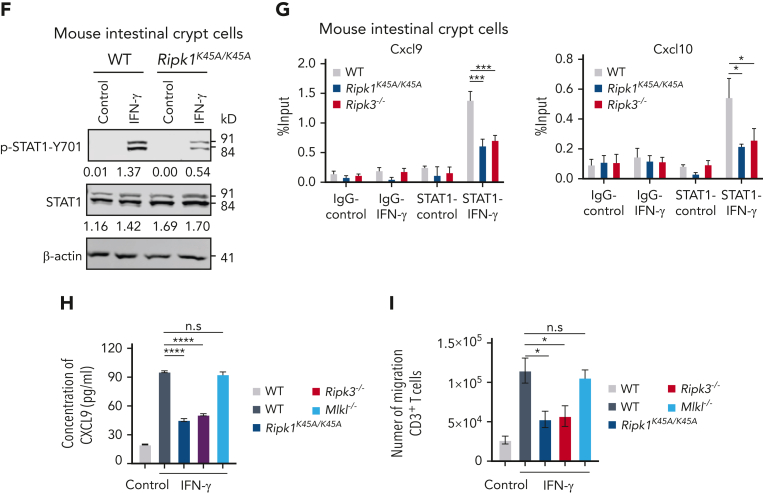
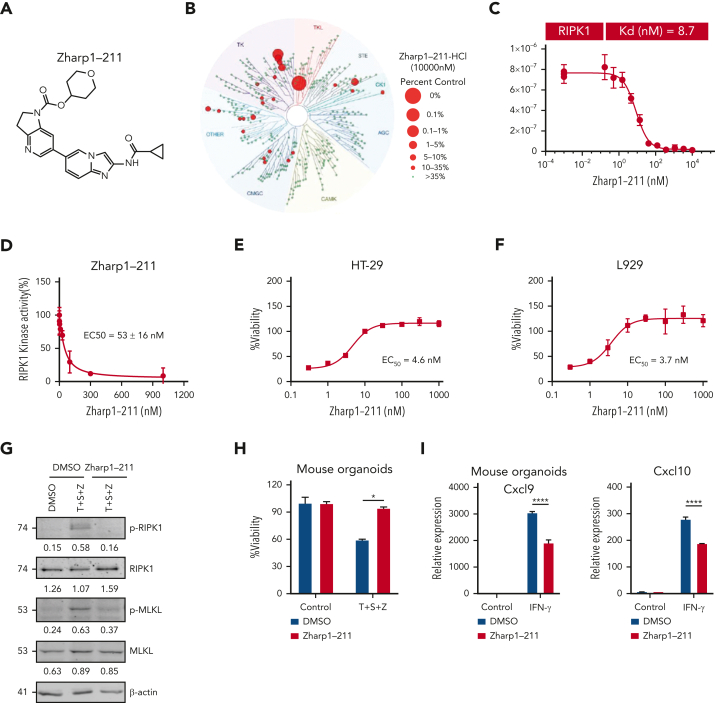
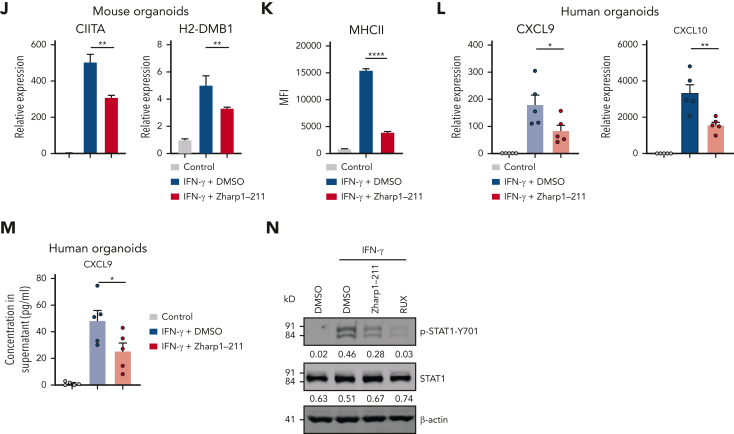
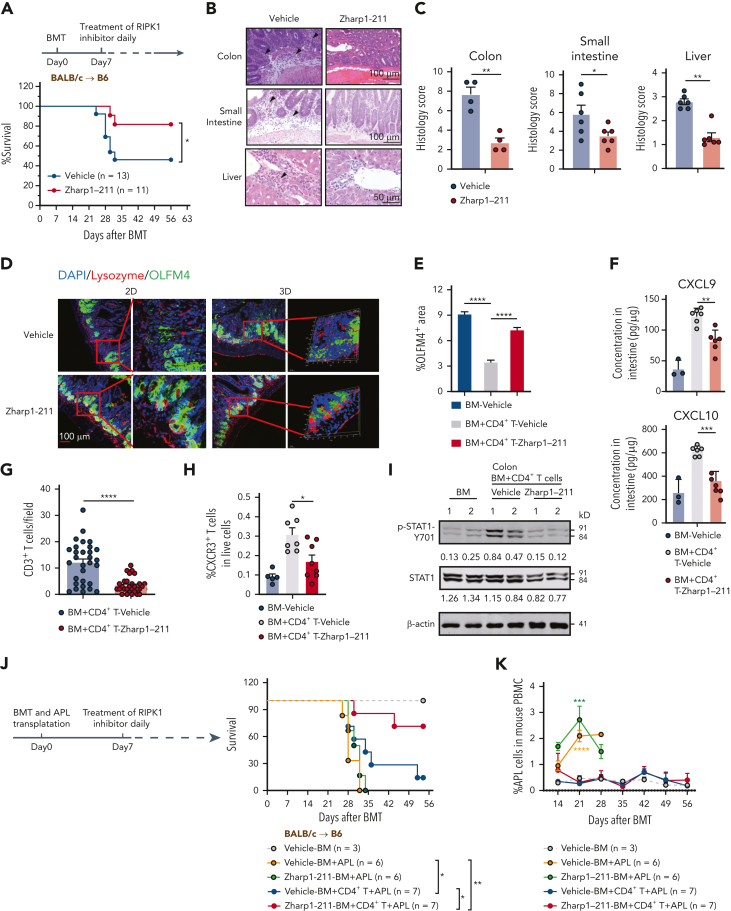
Intestinal epithelial cells (IECs) are implicated in the propagation of T-cell-mediated inflammatory diseases, including graft-versus-host disease (GVHD), but the underlying mechanism remains poorly defined. Here, we report that IECs require receptor-interacting protein kinase-3 (RIPK3) to drive both gastrointestinal (GI) tract and systemic GVHD after allogeneic hematopoietic stem cell transplantation. Selectively inhibiting RIPK3 in IECs markedly reduces GVHD in murine intestine and liver. IEC RIPK3 cooperates with RIPK1 to trigger mixed lineage kinase domain-like protein-independent production of T-cell-recruiting chemokines and major histocompatibility complex (MHC) class II molecules, which amplify and sustain alloreactive T-cell responses. Alloreactive T-cell-produced interferon gamma enhances this RIPK1/RIPK3 action in IECs through a JAK/STAT1-dependent mechanism, creating a feed-forward inflammatory cascade. RIPK1/RIPK3 forms a complex with JAK1 to promote STAT1 activation in IECs. The RIPK1/RIPK3-mediated inflammatory cascade of alloreactive T-cell responses results in intestinal tissue damage, converting the local inflammation into a systemic syndrome. Human patients with severe GVHD showed highly activated RIPK1 in the colon epithelium. Finally, we discover a selective and potent RIPK1 inhibitor (Zharp1-211) that significantly reduces JAK/STAT1-mediated expression of chemokines and MHC class II molecules in IECs, restores intestinal homeostasis, and arrests GVHD without compromising the graft-versus-leukemia (GVL) effect. Thus, targeting RIPK1/RIPK3 in IECs represents an effective nonimmunosuppressive strategy for GVHD treatment and potentially for other diseases involving GI tract inflammation.
© 2023 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: X. Zhang and S. He are co-founders, consultants, and shareholders of Accro Bioscience Inc, which supports research in their labs. The remaining authors declare no competing financial interests.
Figures
Comment in
-
A novel RIPK1 inhibitor attenuates GVHD.Blood. 2023 Mar 2;141(9):969-970. doi: 10.1182/blood.2022019019. Blood. 2023. PMID: 36862437 Free PMC article. No abstract available.
References
-
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14(3):141–153. - PubMed
-
- Patankar JV, Becker C. Cell death in the gut epithelium and implications for chronic inflammation. Nat Rev Gastroenterol Hepatol. 2020;17(9):543–556. - PubMed
-
- Koyama M, Kuns RD, Olver SD, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med. 2011;18(1):135–142. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
