

De novo identification of microbial contaminants in low microbial biomass microbiomes with Squeegee
- PMID: 36357382
- PMCID: PMC9649624
- DOI: 10.1038/s41467-022-34409-z
De novo identification of microbial contaminants in low microbial biomass microbiomes with Squeegee
Abstract
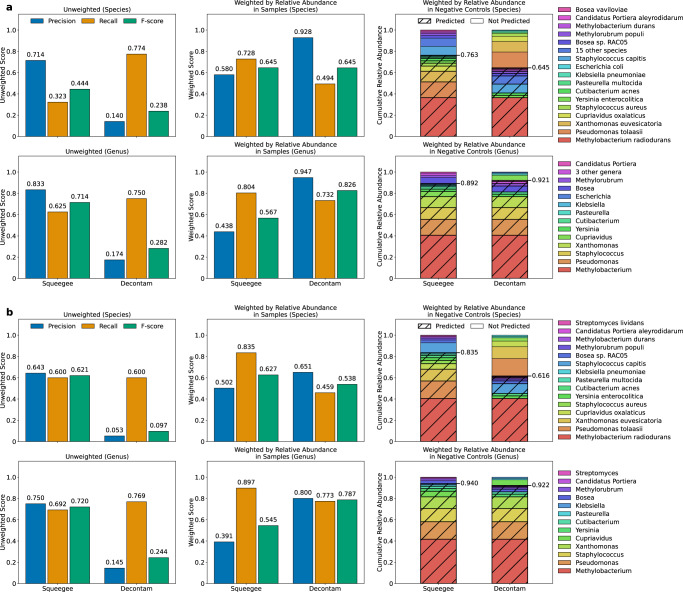
Computational analysis of host-associated microbiomes has opened the door to numerous discoveries relevant to human health and disease. However, contaminant sequences in metagenomic samples can potentially impact the interpretation of findings reported in microbiome studies, especially in low-biomass environments. Contamination from DNA extraction kits or sampling lab environments leaves taxonomic "bread crumbs" across multiple distinct sample types. Here we describe Squeegee, a de novo contamination detection tool that is based upon this principle, allowing the detection of microbial contaminants when negative controls are unavailable. On the low-biomass samples, we compare Squeegee predictions to experimental negative control data and show that Squeegee accurately recovers putative contaminants. We analyze samples of varying biomass from the Human Microbiome Project and identify likely, previously unreported kit contamination. Collectively, our results highlight that Squeegee can identify microbial contaminants with high precision and thus represents a computational approach for contaminant detection when negative controls are unavailable.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





Similar articles
-
The impact of kit, environment, and sampling contamination on the observed microbiome of bovine milk.mSystems. 2024 Jun 18;9(6):e0115823. doi: 10.1128/msystems.01158-23. Epub 2024 May 24. mSystems. 2024. PMID: 38785438 Free PMC article.
-
Contamination in Low Microbial Biomass Microbiome Studies: Issues and Recommendations.Trends Microbiol. 2019 Feb;27(2):105-117. doi: 10.1016/j.tim.2018.11.003. Epub 2018 Nov 26. Trends Microbiol. 2019. PMID: 30497919 Review.
-
Recentrifuge: Robust comparative analysis and contamination removal for metagenomics.PLoS Comput Biol. 2019 Apr 8;15(4):e1006967. doi: 10.1371/journal.pcbi.1006967. eCollection 2019 Apr. PLoS Comput Biol. 2019. PMID: 30958827 Free PMC article.
-
Impact of DNA extraction, sample dilution, and reagent contamination on 16S rRNA gene sequencing of human feces.Appl Microbiol Biotechnol. 2018 Jan;102(1):403-411. doi: 10.1007/s00253-017-8583-z. Epub 2017 Oct 27. Appl Microbiol Biotechnol. 2018. PMID: 29079861
-
Practical considerations for sampling and data analysis in contemporary metagenomics-based environmental studies.J Microbiol Methods. 2018 Nov;154:14-18. doi: 10.1016/j.mimet.2018.09.020. Epub 2018 Oct 1. J Microbiol Methods. 2018. PMID: 30287354 Review.
Cited by
-
AMAnD: an automated metagenome anomaly detection methodology utilizing DeepSVDD neural networks.Front Public Health. 2023 Jul 11;11:1181911. doi: 10.3389/fpubh.2023.1181911. eCollection 2023. Front Public Health. 2023. PMID: 37497030 Free PMC article.
-
A brain microbiome in salmonids at homeostasis.Sci Adv. 2024 Sep 20;10(38):eado0277. doi: 10.1126/sciadv.ado0277. Epub 2024 Sep 18. Sci Adv. 2024. PMID: 39292785 Free PMC article.
-
Guidelines for preventing and reporting contamination in low-biomass microbiome studies.Nat Microbiol. 2025 Jul;10(7):1570-1580. doi: 10.1038/s41564-025-02035-2. Epub 2025 Jun 20. Nat Microbiol. 2025. PMID: 40542287 Review.
-
The Skin Microbiome: Current Techniques, Challenges, and Future Directions.Microorganisms. 2023 May 6;11(5):1222. doi: 10.3390/microorganisms11051222. Microorganisms. 2023. PMID: 37317196 Free PMC article. Review.
-
Multi-kingdom microbiota analysis reveals bacteria-viral interplay in IBS with depression and anxiety.NPJ Biofilms Microbiomes. 2025 Jul 5;11(1):129. doi: 10.1038/s41522-025-00760-4. NPJ Biofilms Microbiomes. 2025. PMID: 40617850 Free PMC article.
References
-
- Fox Gc-a, et al. The phylogeny of prokaryotes. Science. 1980;209:457–463. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
