

Rare and common genetic determinants of metabolic individuality and their effects on human health
- PMID: 36357675
- PMCID: PMC9671801
- DOI: 10.1038/s41591-022-02046-0
Rare and common genetic determinants of metabolic individuality and their effects on human health
Abstract
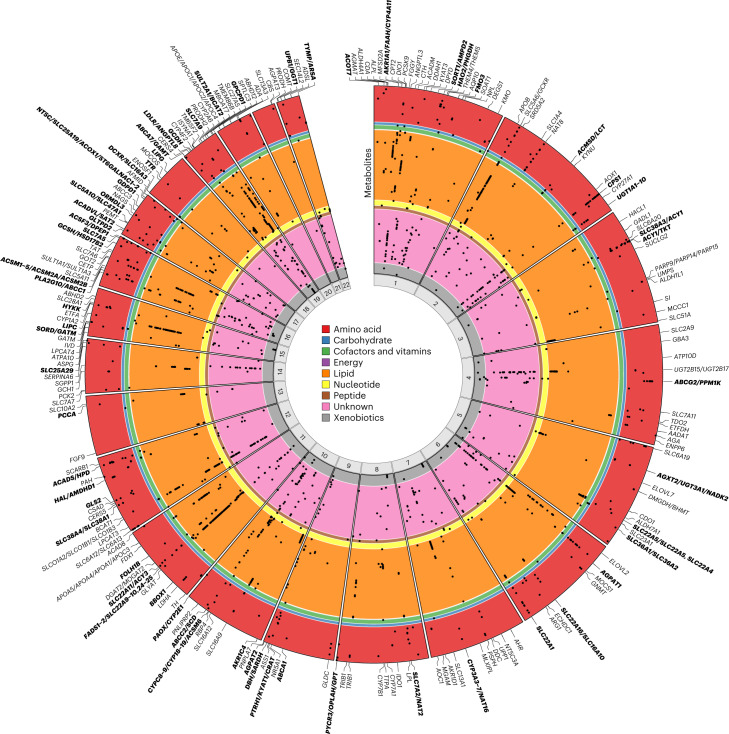
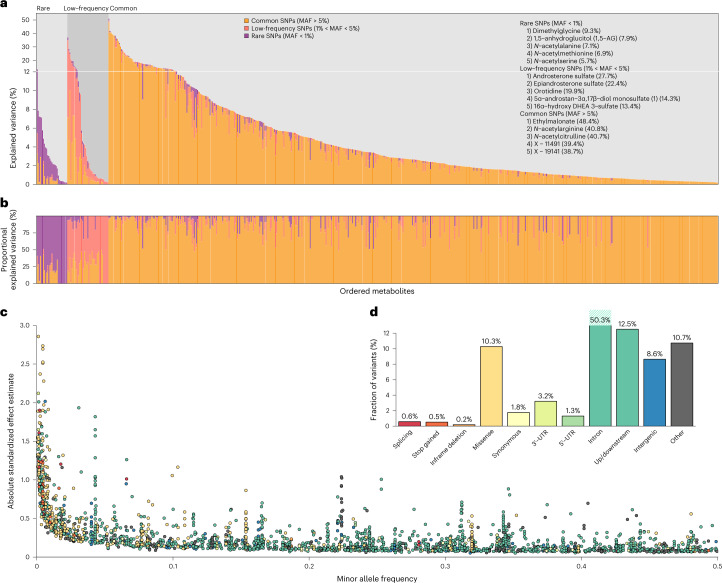
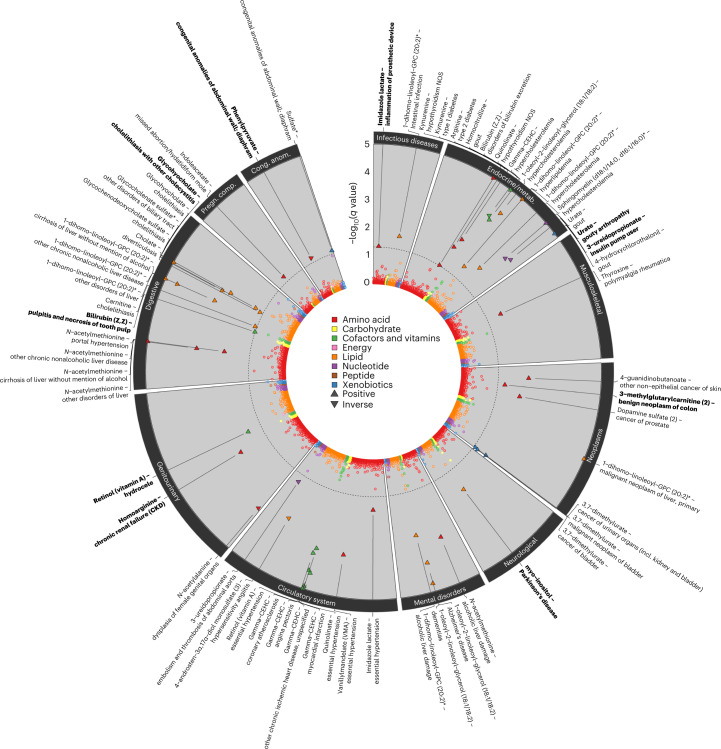
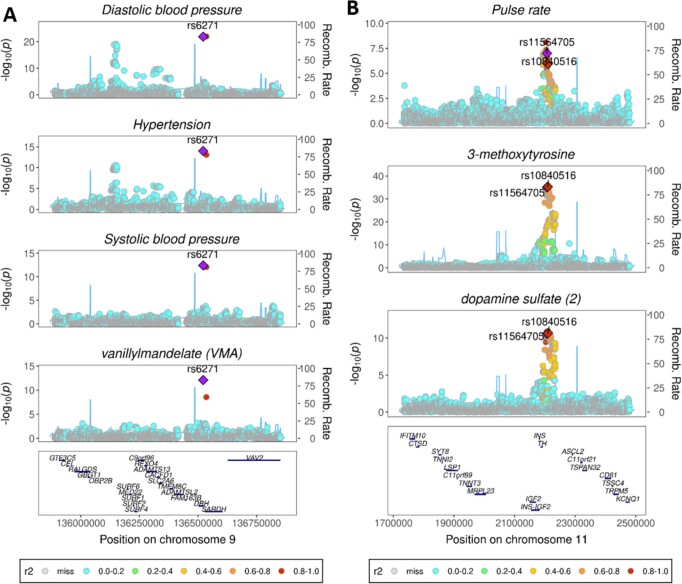
Garrod's concept of 'chemical individuality' has contributed to comprehension of the molecular origins of human diseases. Untargeted high-throughput metabolomic technologies provide an in-depth snapshot of human metabolism at scale. We studied the genetic architecture of the human plasma metabolome using 913 metabolites assayed in 19,994 individuals and identified 2,599 variant-metabolite associations (P < 1.25 × 10-11) within 330 genomic regions, with rare variants (minor allele frequency ≤ 1%) explaining 9.4% of associations. Jointly modeling metabolites in each region, we identified 423 regional, co-regulated, variant-metabolite clusters called genetically influenced metabotypes. We assigned causal genes for 62.4% of these genetically influenced metabotypes, providing new insights into fundamental metabolite physiology and clinical relevance, including metabolite-guided discovery of potential adverse drug effects (DPYD and SRD5A2). We show strong enrichment of inborn errors of metabolism-causing genes, with examples of metabolite associations and clinical phenotypes of non-pathogenic variant carriers matching characteristics of the inborn errors of metabolism. Systematic, phenotypic follow-up of metabolite-specific genetic scores revealed multiple potential etiological relationships.
© 2022. The Author(s).
Conflict of interest statement
During the course of the project, P.S. became a full-time employee of GlaxoSmithKline, V.P.W.A. became a full-time employee of AstraZeneca, L.B. became a full-time employee of BioMarin, J.Z. became a full-time employee of Novartis, J.M.M.H. became a full-time employee of Novo Nordisk Ltd. and L.A.L. is presently an employee and owns stocks and stock options of Regeneron Pharmaceuticals Inc. E.R.G. receives an honorarium from the journal
Figures









Comment in
-
Metabolic individuality: Limitations, challenges, and potential for clinical utility.Cell Metab. 2023 Feb 7;35(2):233-235. doi: 10.1016/j.cmet.2023.01.007. Cell Metab. 2023. PMID: 36754017
References
-
- Nag, A. et al. Assessing the contribution of rare-to-common protein-coding variants to circulating metabolic biomarker levels via 412,394 UK Biobank exome sequences. Preprint at medRxiv10.1101/2021.12.24.21268381 (2021).
-
- Long, T. et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 10.1038/ng.3809 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 HG011138/HG/NHGRI NIH HHS/United States
- BTRU-2014-10024/DH_/Department of Health/United Kingdom
- 206194/WT_/Wellcome Trust/United Kingdom
- MC_UU_12015/1/MRC_/Medical Research Council/United Kingdom
- WT209492/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- CH/12/2/29428/BHF_/British Heart Foundation/United Kingdom
- BCDSA\100005/BHF_/British Heart Foundation/United Kingdom
- RF1 AG059093/AG/NIA NIH HHS/United States
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
- RG/13/13/30194/BHF_/British Heart Foundation/United Kingdom
- MC_UU_00006/1/MRC_/Medical Research Council/United Kingdom
- MR/L003120/1/MRC_/Medical Research Council/United Kingdom
- R01 GM140287/GM/NIGMS NIH HHS/United States
- MR/S003746/1/MRC_/Medical Research Council/United Kingdom
- BRC-1215-20014/DH_/Department of Health/United Kingdom
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- MC-UU_12015/1/MRC_/Medical Research Council/United Kingdom
- RG/18/13/33946/BHF_/British Heart Foundation/United Kingdom
- BRC-1215-20009/DH_/Department of Health/United Kingdom
- G1000143/MRC_/Medical Research Council/United Kingdom
- U19 AG063744/AG/NIA NIH HHS/United States
- C864/A14136/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_00006/1 /MRC_/Medical Research Council/United Kingdom
- MR/N003284/1/MRC_/Medical Research Council/United Kingdom
- 14136/CRUK_/Cancer Research UK/United Kingdom
- 221651/Z/20/Z/WT_/Wellcome Trust/United Kingdom
- AA/18/6/24223/BHF_/British Heart Foundation/United Kingdom
- G0401527/MRC_/Medical Research Council/United Kingdom
- MR/L00002/1/MRC_/Medical Research Council/United Kingdom
- 203810/Z/16/A/WT_/Wellcome Trust/United Kingdom
- U01 AG061359/AG/NIA NIH HHS/United States
- RF1 AG057452/AG/NIA NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R56 AG068026/AG/NIA NIH HHS/United States
- MC_PC_13048/MRC_/Medical Research Council/United Kingdom
- SP/09/002/BHF_/British Heart Foundation/United Kingdom
- R35 HG010718/HG/NHGRI NIH HHS/United States
