

Divergent SARS-CoV-2 variant emerges in white-tailed deer with deer-to-human transmission
- PMID: 36357713
- PMCID: PMC9712111
- DOI: 10.1038/s41564-022-01268-9
Divergent SARS-CoV-2 variant emerges in white-tailed deer with deer-to-human transmission
Erratum in
-
Publisher Correction: Divergent SARS-CoV-2 variant emerges in white-tailed deer with deer-to-human transmission.Nat Microbiol. 2023 Jan;8(1):188. doi: 10.1038/s41564-022-01298-3. Nat Microbiol. 2023. PMID: 36509933 Free PMC article. No abstract available.
Abstract
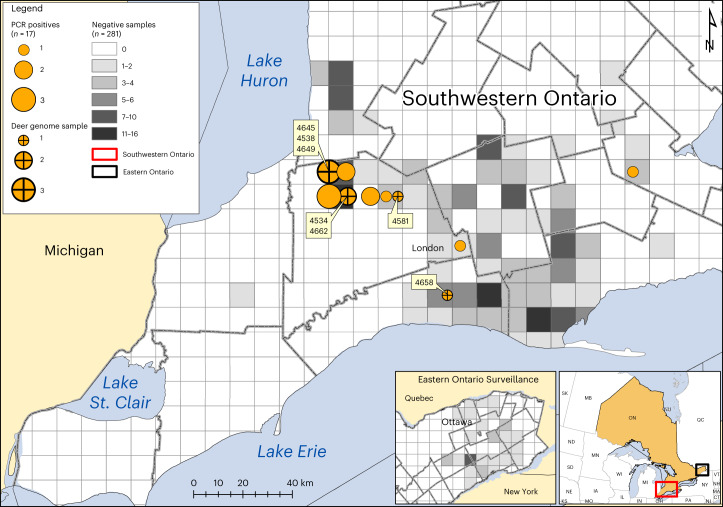
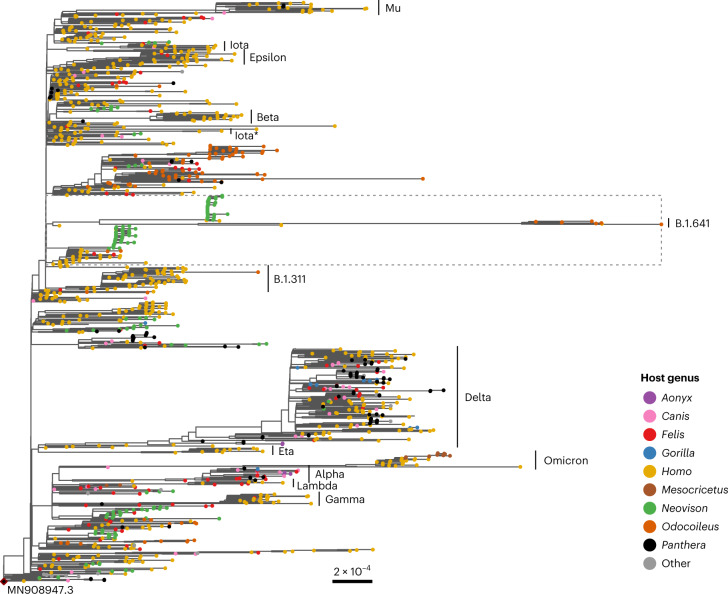
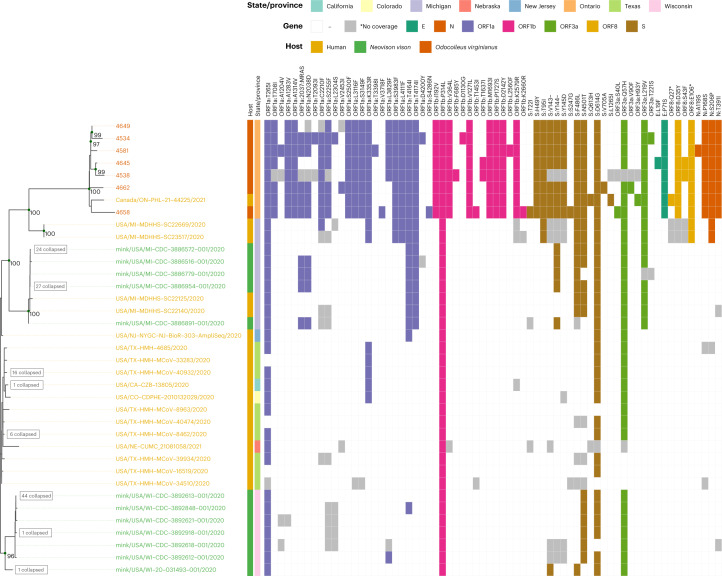
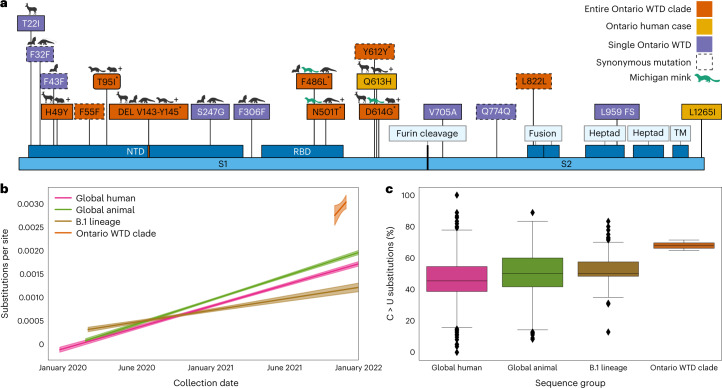
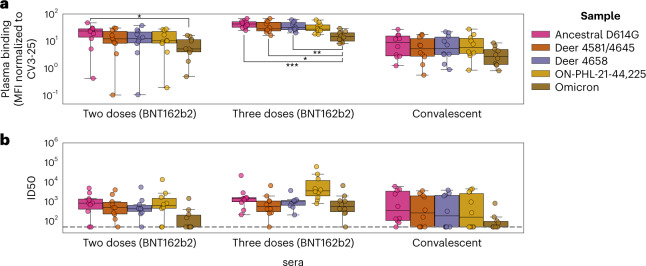
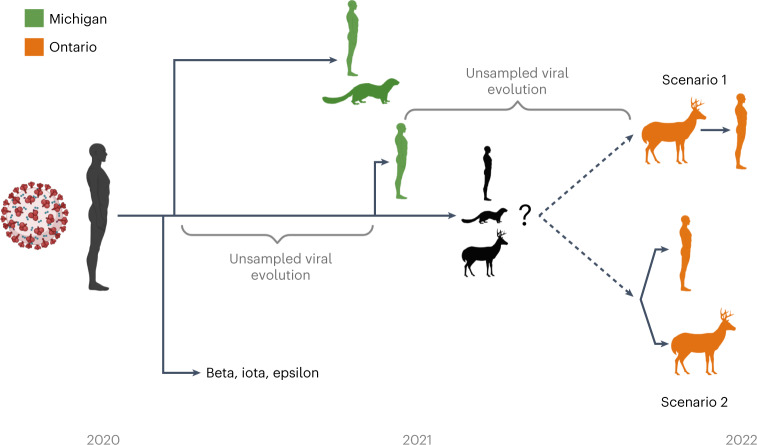
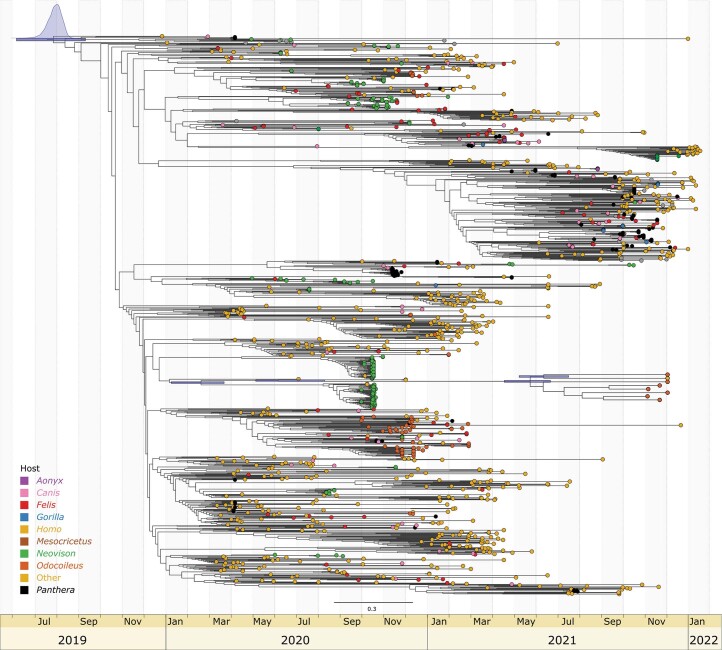
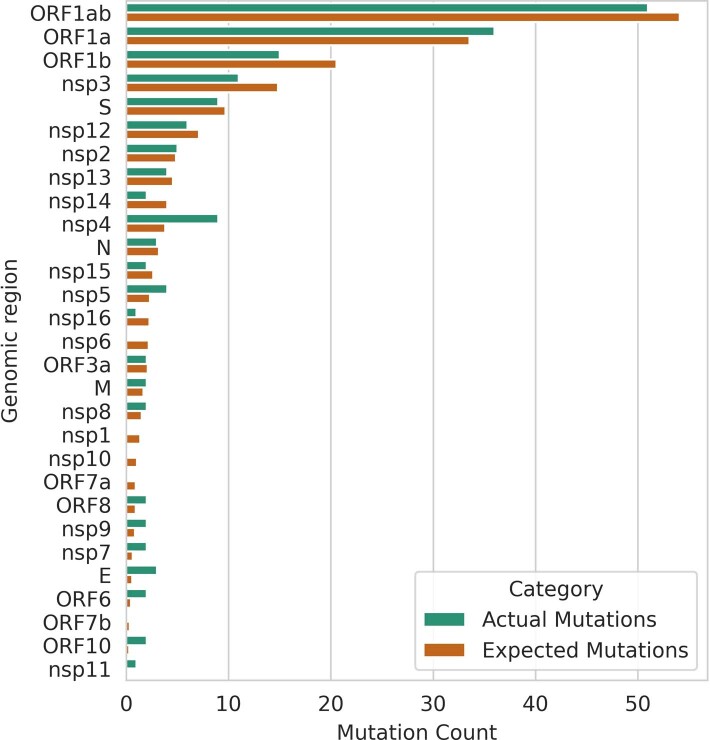
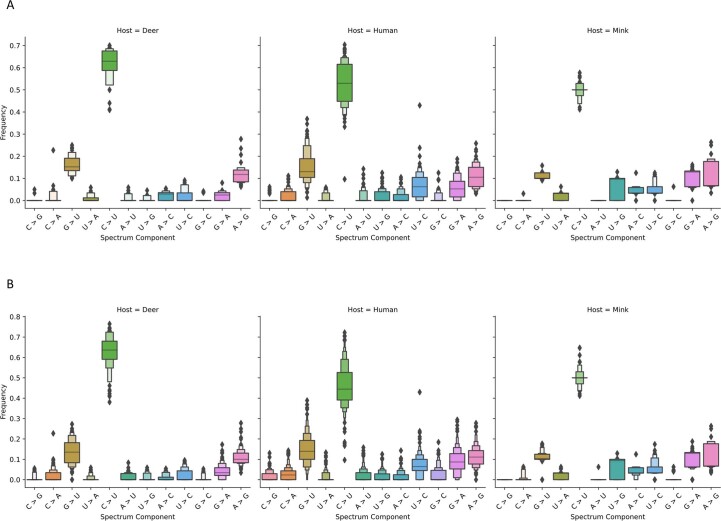
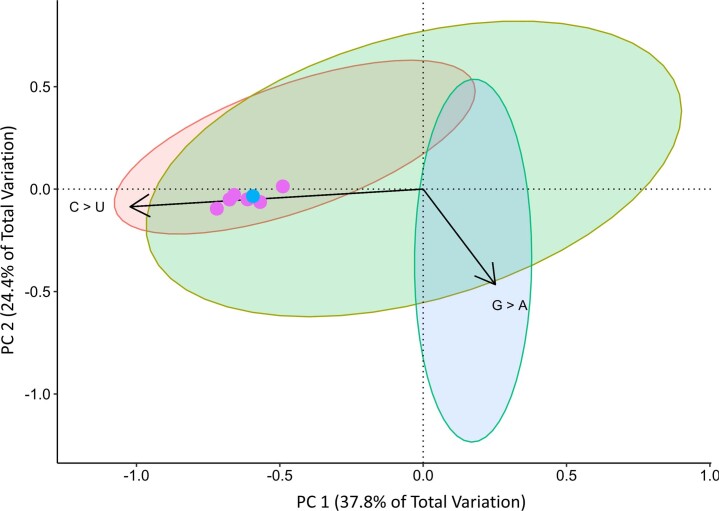
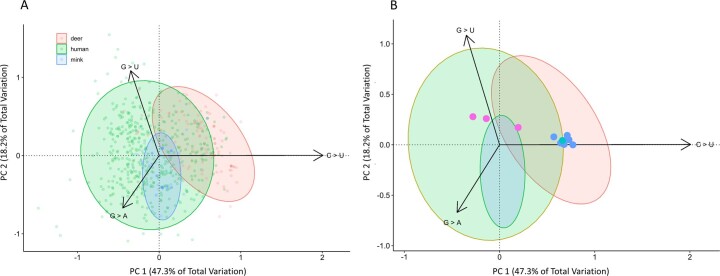
Wildlife reservoirs of broad-host-range viruses have the potential to enable evolution of viral variants that can emerge to infect humans. In North America, there is phylogenomic evidence of continual transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from humans to white-tailed deer (Odocoileus virginianus) through unknown means, but no evidence of transmission from deer to humans. We carried out an observational surveillance study in Ontario, Canada during November and December 2021 (n = 300 deer) and identified a highly divergent lineage of SARS-CoV-2 in white-tailed deer (B.1.641). This lineage is one of the most divergent SARS-CoV-2 lineages identified so far, with 76 mutations (including 37 previously associated with non-human mammalian hosts). From a set of five complete and two partial deer-derived viral genomes we applied phylogenomic, recombination, selection and mutation spectrum analyses, which provided evidence for evolution and transmission in deer and a shared ancestry with mink-derived virus. Our analysis also revealed an epidemiologically linked human infection. Taken together, our findings provide evidence for sustained evolution of SARS-CoV-2 in white-tailed deer and of deer-to-human transmission.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Viral spillback.Nat Rev Microbiol. 2023 Jan;21(1):2. doi: 10.1038/s41579-022-00829-3. Nat Rev Microbiol. 2023. PMID: 36400847 Free PMC article.
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
