

The DNA Methylation in Neurological Diseases
- PMID: 36359835
- PMCID: PMC9657829
- DOI: 10.3390/cells11213439
The DNA Methylation in Neurological Diseases
Abstract
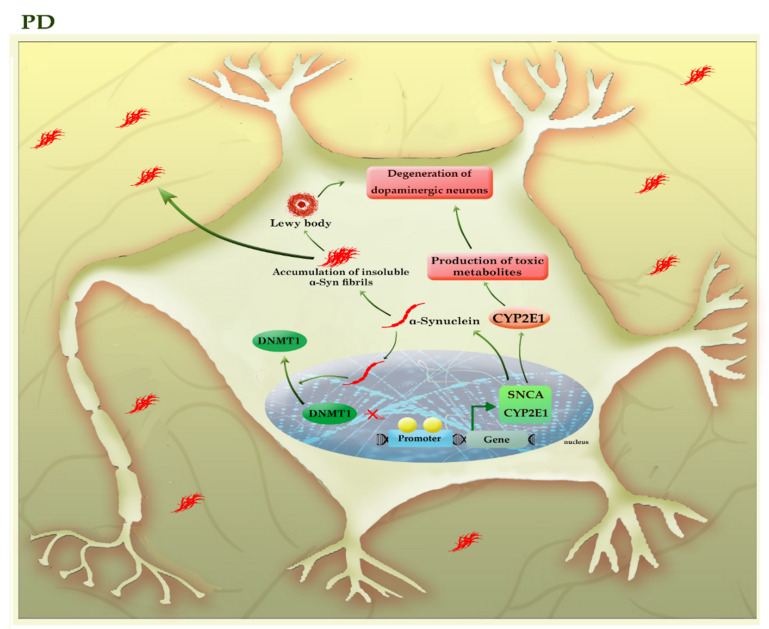
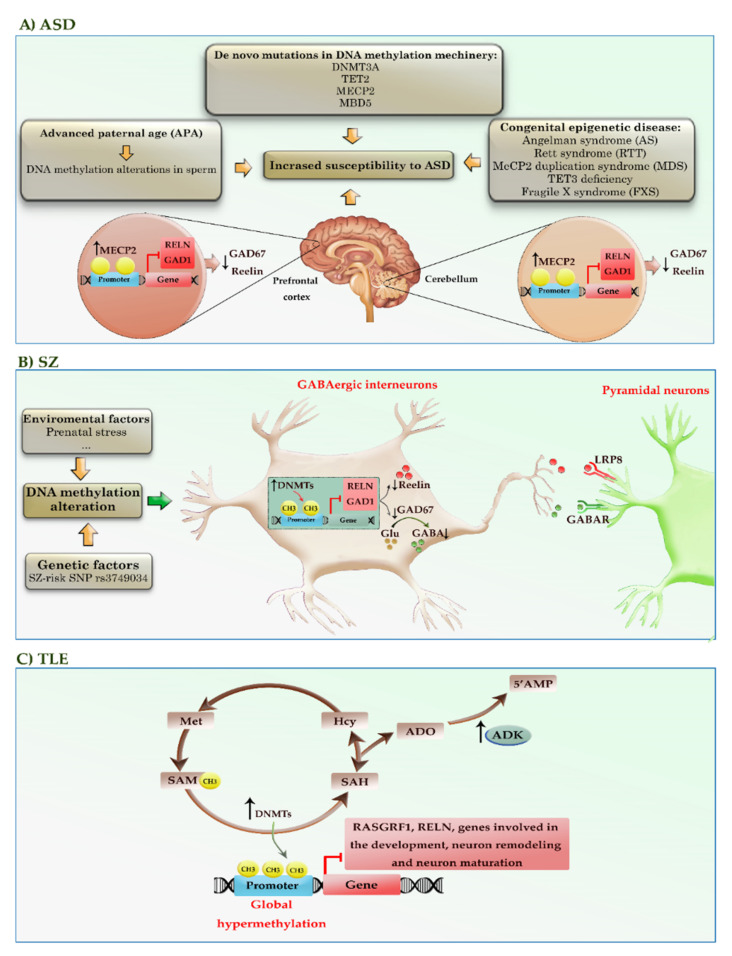
DNA methylation is critical for the normal development and functioning of the human brain, such as the proliferation and differentiation of neural stem cells, synaptic plasticity, neuronal reparation, learning, and memory. Despite the physical stability of DNA and methylated DNA compared to other epigenetic modifications, some DNA methylation-based biomarkers have translated into clinical practice. Increasing reports indicate a strong association between DNA methylation profiles and various clinical outcomes in neurological diseases, making DNA methylation profiles valuable as novel clinical markers. In this review, we aim to discuss the latest evidence concerning DNA methylation alterations in the development of neurodegenerative, neurodevelopmental, and neuropsychiatric diseases. We also highlighted the relationship of DNA methylation alterations with the disease progression and outcome in many neurological diseases such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, frontotemporal dementia, and autism.
Keywords: Alzheimer’s disease; DNA methylation; Huntington’s disease; Parkinson’s disease; autism; neurological disorders.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
