

SIRT1 Promotes Host Protective Immunity against Toxoplasma gondii by Controlling the FoxO-Autophagy Axis via the AMPK and PI3K/AKT Signalling Pathways
- PMID: 36362370
- PMCID: PMC9654124
- DOI: 10.3390/ijms232113578
SIRT1 Promotes Host Protective Immunity against Toxoplasma gondii by Controlling the FoxO-Autophagy Axis via the AMPK and PI3K/AKT Signalling Pathways
Abstract
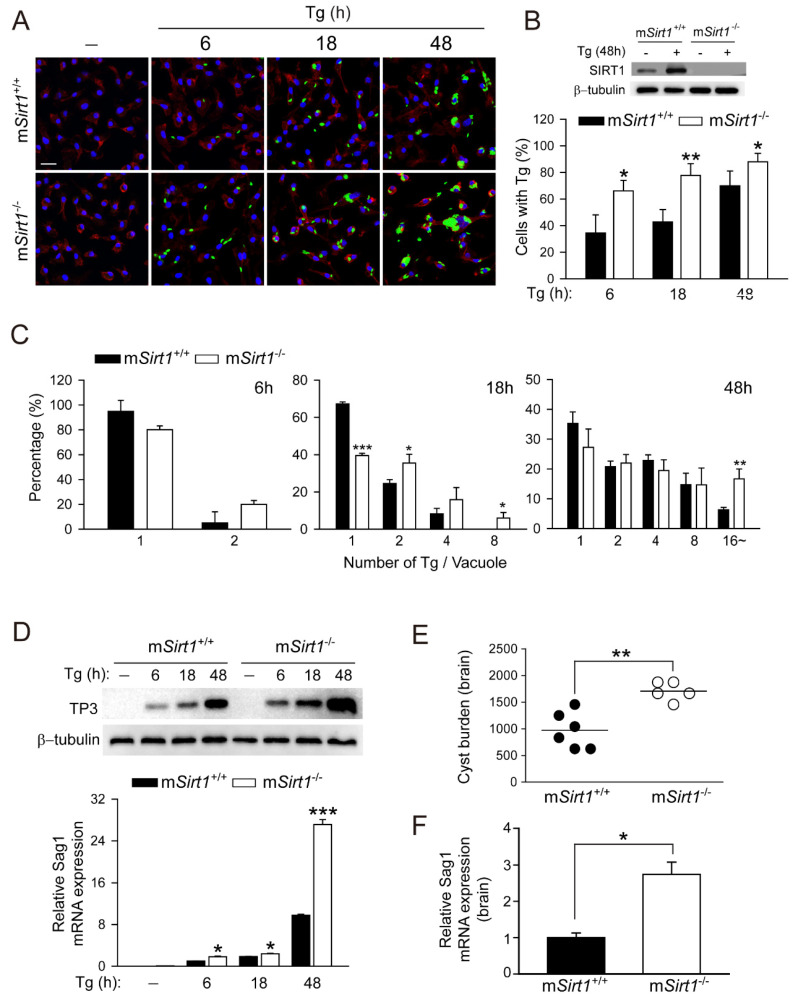
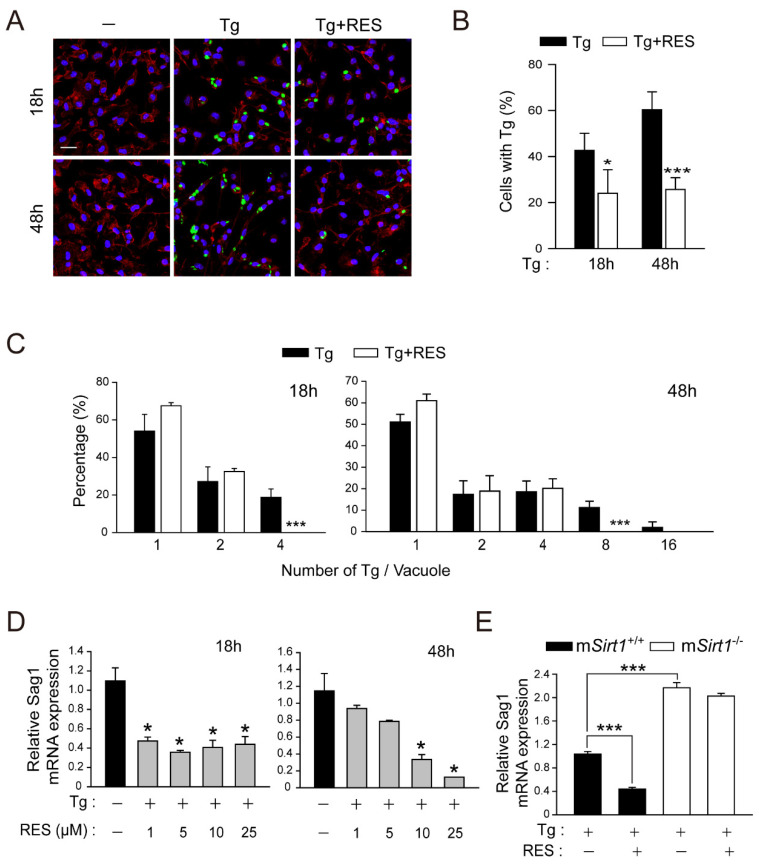
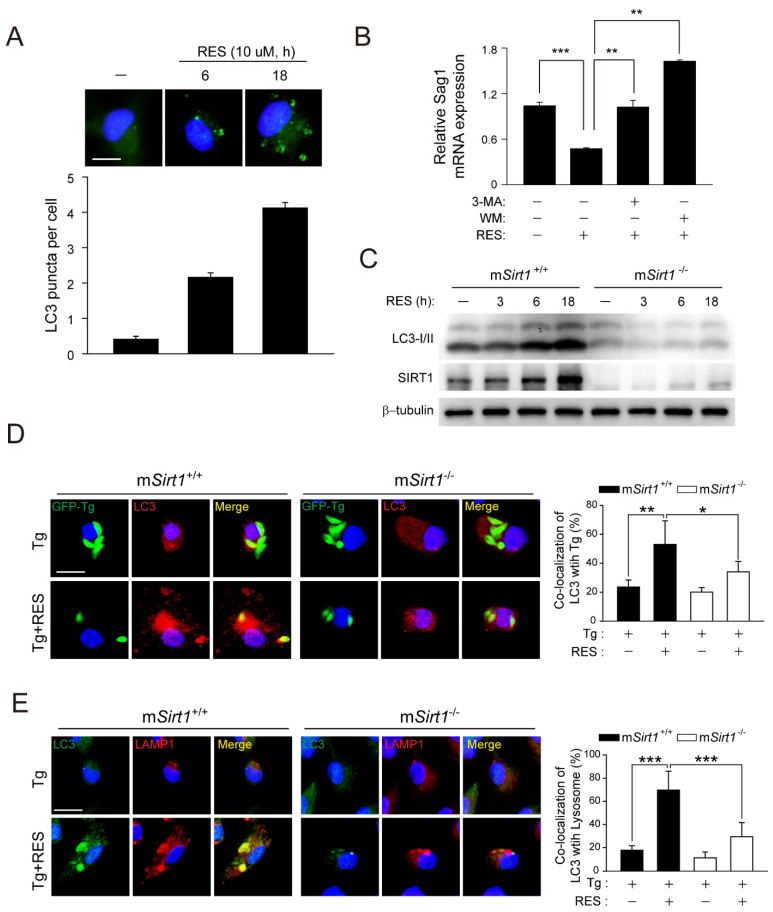
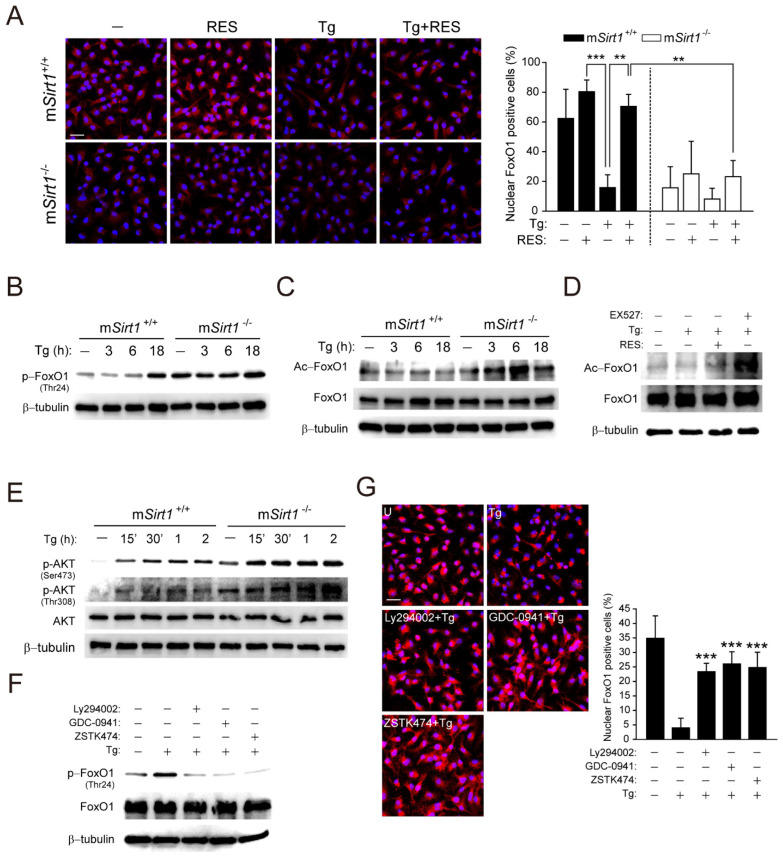
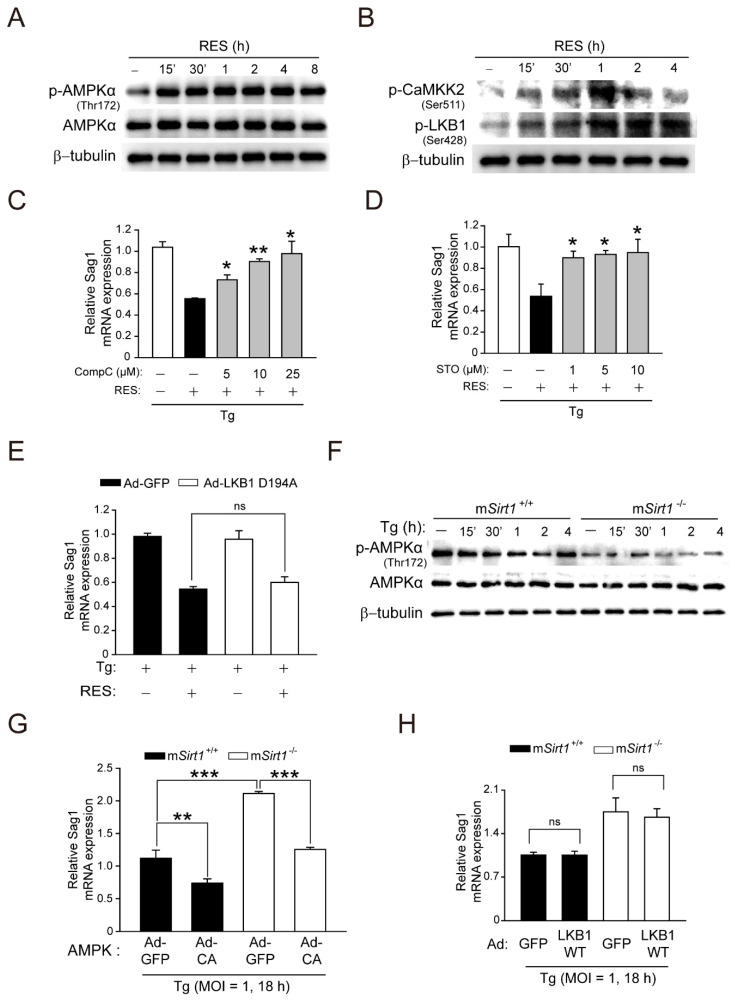
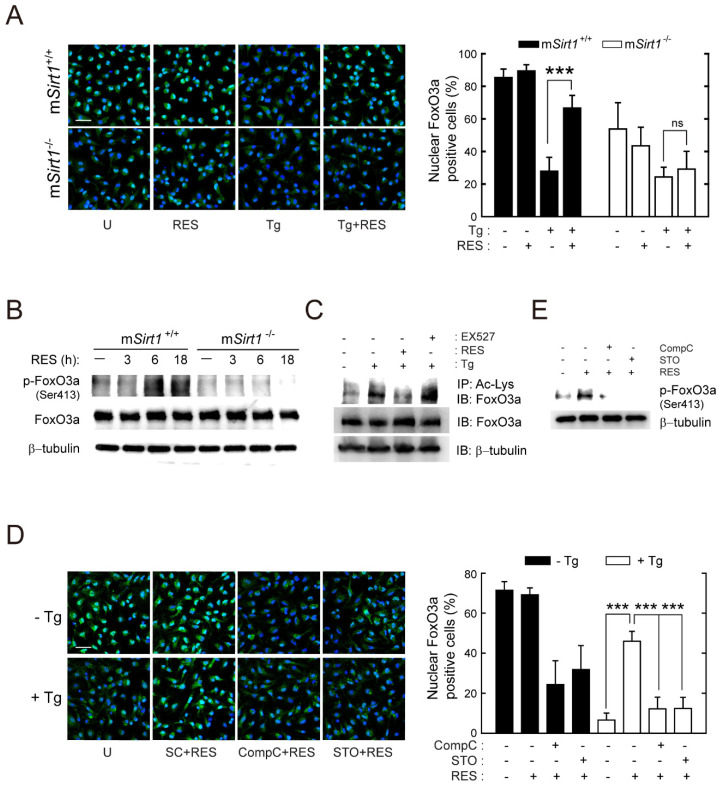
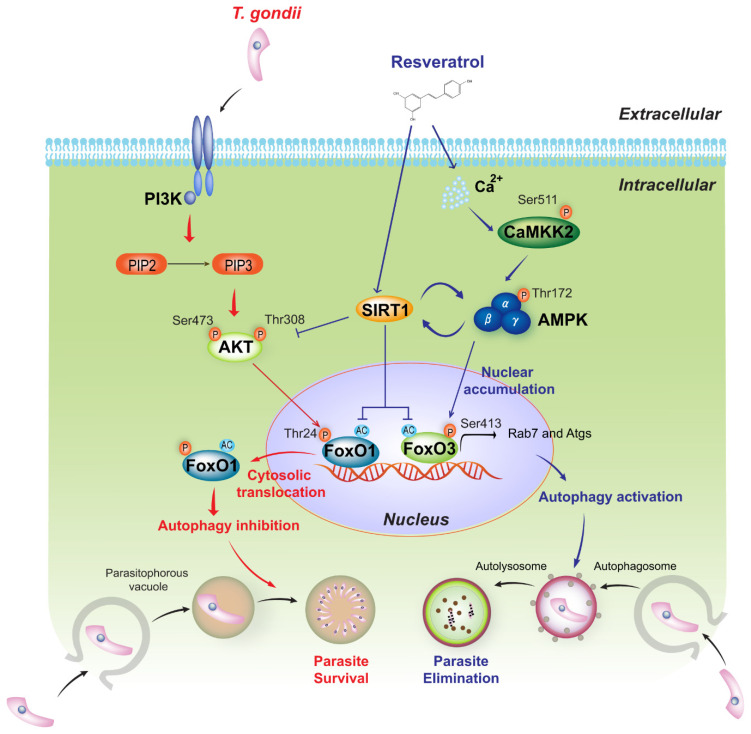
Sirtuin 1 (SIRT1) regulates cellular processes by deacetylating non-histone targets, including transcription factors and intracellular signalling mediators; thus, its abnormal activation is closely linked to the pathophysiology of several diseases. However, its function in Toxoplasma gondii infection is unclear. We found that SIRT1 contributes to autophagy activation via the AMP-activated protein kinase (AMPK) and PI3K/AKT signalling pathways, promoting anti-Toxoplasma responses. Myeloid-specific Sirt1-/- mice exhibited an increased cyst burden in brain tissue compared to wild-type mice following infection with the avirulent ME49 strain. Consistently, the intracellular survival of T. gondii was markedly increased in Sirt1-deficient bone-marrow-derived macrophages (BMDMs). In contrast, the activation of SIRT1 by resveratrol resulted in not only the induction of autophagy but also a significantly increased anti-Toxoplasma effect. Notably, SIRT1 regulates the FoxO-autophagy axis in several human diseases. Importantly, the T. gondii-induced phosphorylation, acetylation, and cytosolic translocation of FoxO1 was enhanced in Sirt1-deficient BMDMs and the pharmacological inhibition of PI3K/AKT signalling reduced the cytosolic translocation of FoxO1 in BMDMs infected with T. gondii. Further, the CaMKK2-dependent AMPK signalling pathway is responsible for the effect of SIRT1 on the FoxO3a-autophagy axis and for its anti-Toxoplasma activity. Collectively, our findings reveal a previously unappreciated role for SIRT1 in Toxoplasma infection.
Keywords: AMP-activated protein kinase; Class O of forkhead box transcription factors; PI3K/AKT signalling pathway; Sirtuin 1; Toxoplasma gondii; autophagy; bone-marrow-derived macrophages.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Luft B.J., Hafner R., Korzun A.H., Leport C., Antoniskis D., Bosler E.M., Bourland D.D., 3rd, Uttamchandani R., Fuhrer J., Jacobson J., et al. Toxoplasmic encephalitis in patients with the acquired immunodeficiency syndrome. Members of the ACTG 077p/ANRS 009 Study Team. N. Engl. J. Med. 1993;329:995–1000. doi: 10.1056/NEJM199309303291403. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
