

The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest
- PMID: 36370105
- PMCID: PMC9825434
- DOI: 10.1093/nar/gkac1000
The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest
Abstract
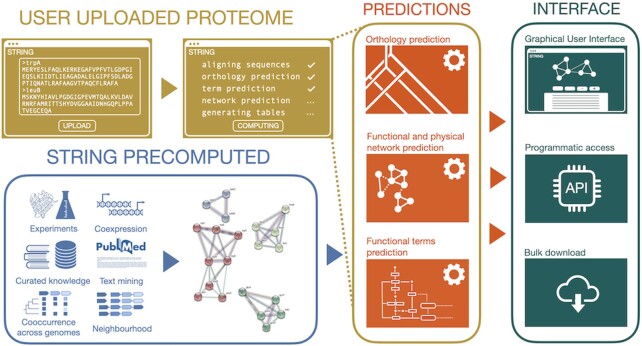
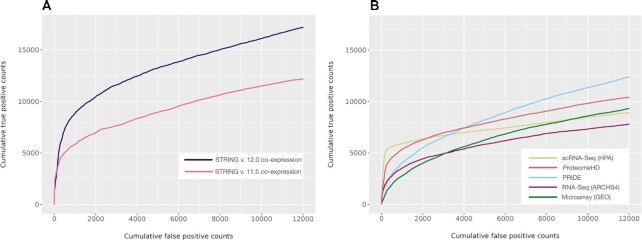
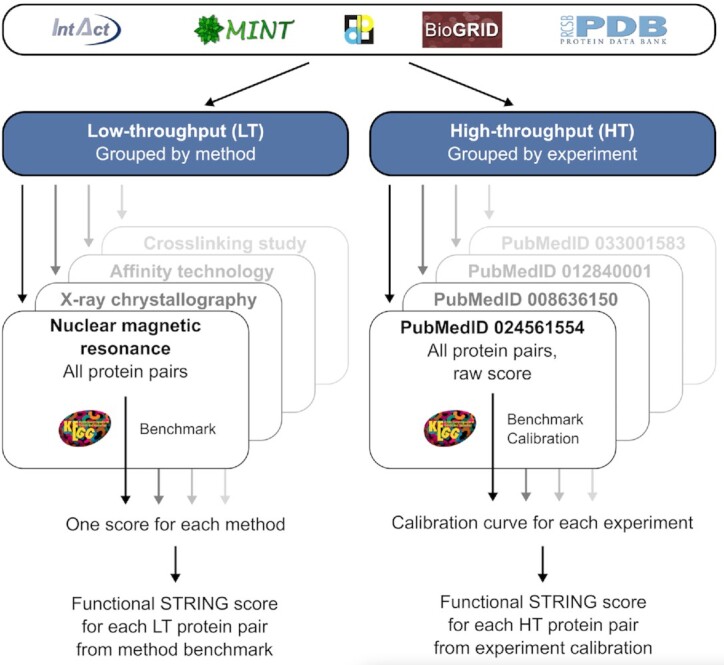
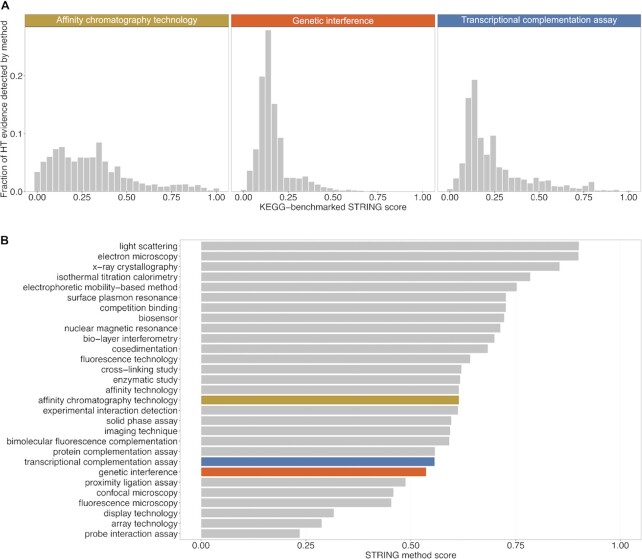
Much of the complexity within cells arises from functional and regulatory interactions among proteins. The core of these interactions is increasingly known, but novel interactions continue to be discovered, and the information remains scattered across different database resources, experimental modalities and levels of mechanistic detail. The STRING database (https://string-db.org/) systematically collects and integrates protein-protein interactions-both physical interactions as well as functional associations. The data originate from a number of sources: automated text mining of the scientific literature, computational interaction predictions from co-expression, conserved genomic context, databases of interaction experiments and known complexes/pathways from curated sources. All of these interactions are critically assessed, scored, and subsequently automatically transferred to less well-studied organisms using hierarchical orthology information. The data can be accessed via the website, but also programmatically and via bulk downloads. The most recent developments in STRING (version 12.0) are: (i) it is now possible to create, browse and analyze a full interaction network for any novel genome of interest, by submitting its complement of encoded proteins, (ii) the co-expression channel now uses variational auto-encoders to predict interactions, and it covers two new sources, single-cell RNA-seq and experimental proteomics data and (iii) the confidence in each experimentally derived interaction is now estimated based on the detection method used, and communicated to the user in the web-interface. Furthermore, STRING continues to enhance its facilities for functional enrichment analysis, which are now fully available also for user-submitted genomes.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Przybyla L., Gilbert L.A.. A new era in functional genomics screens. Nat. Rev. Genet. 2022; 23:89–103. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
