

Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells
- PMID: 36376280
- PMCID: PMC9663558
- DOI: 10.1038/s41467-022-34095-x
Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells
Abstract
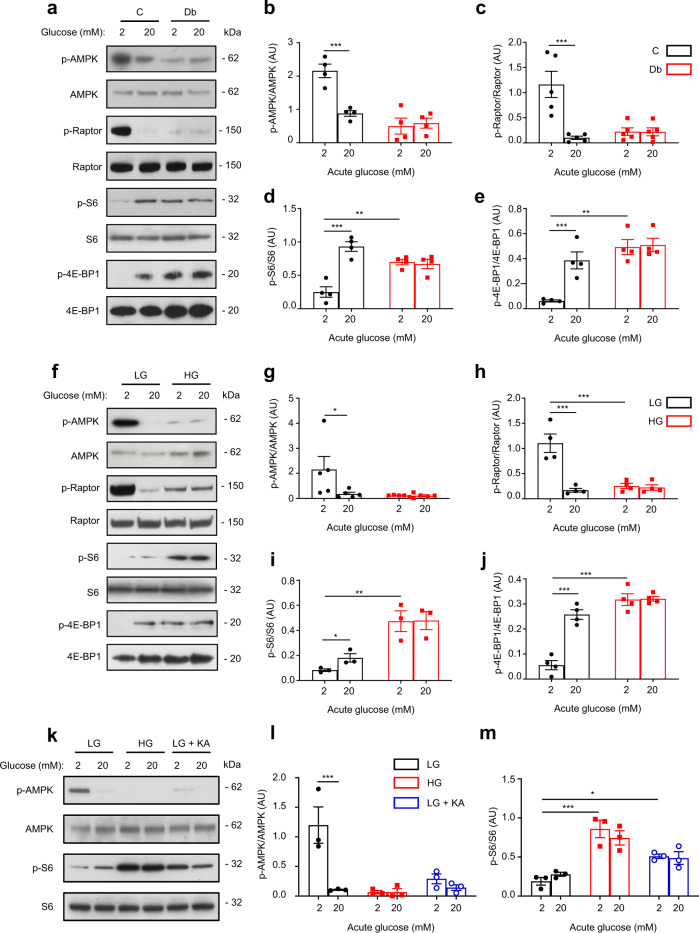
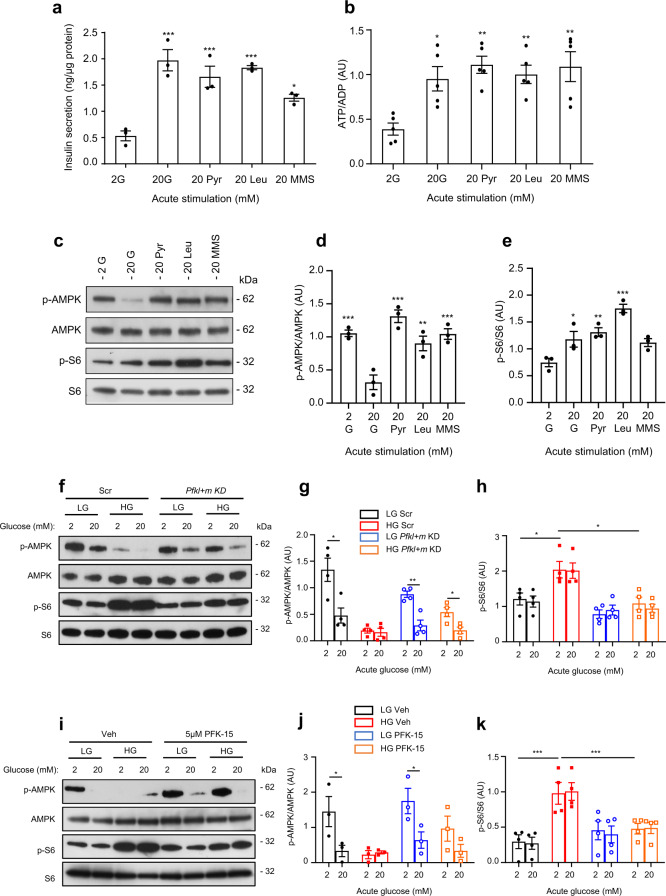
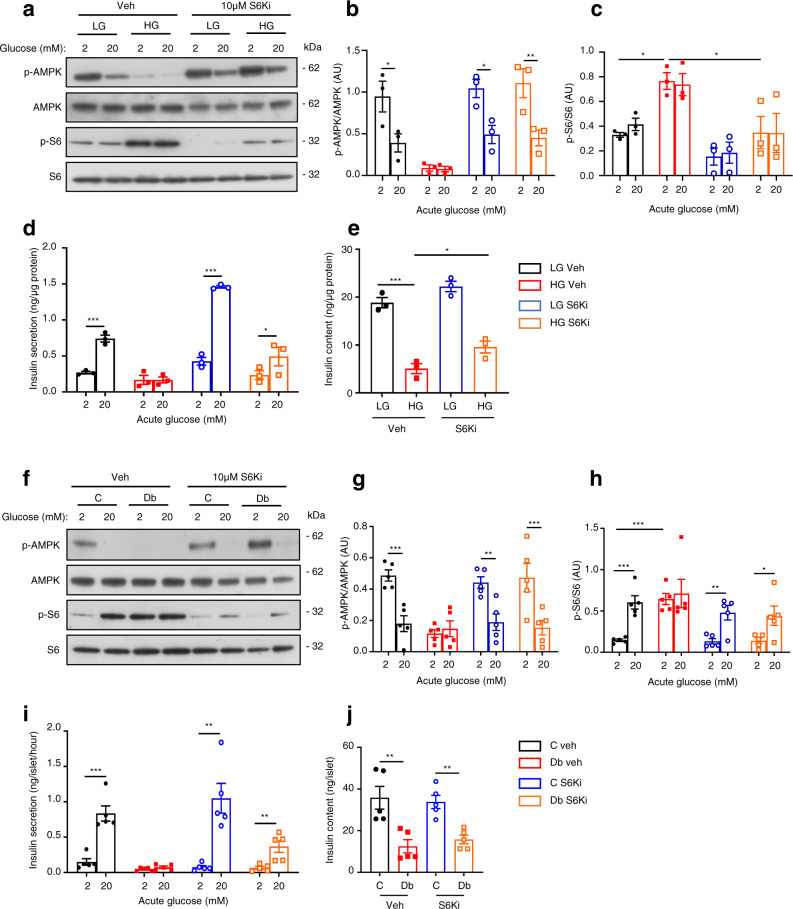
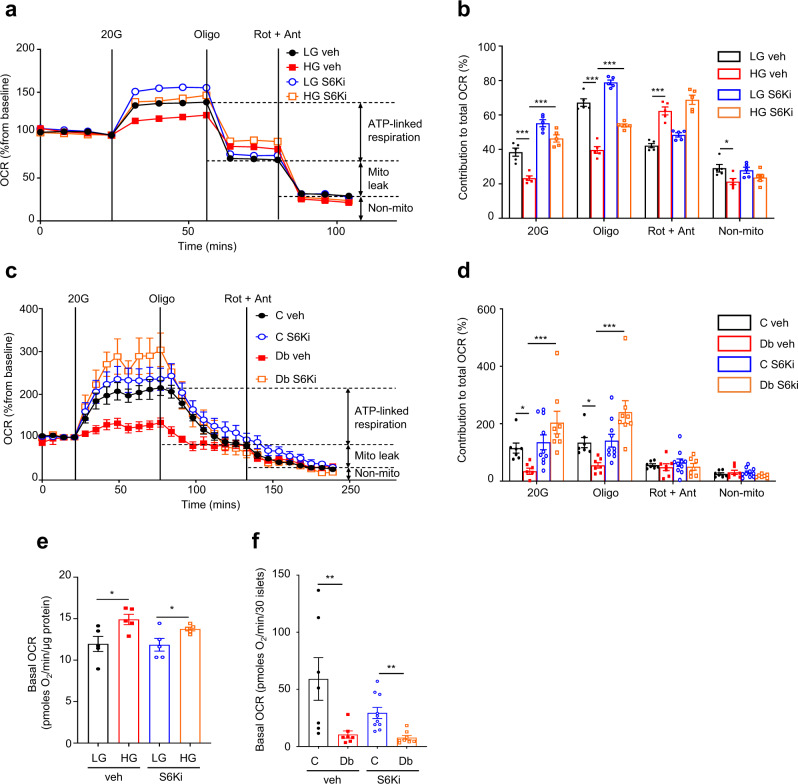
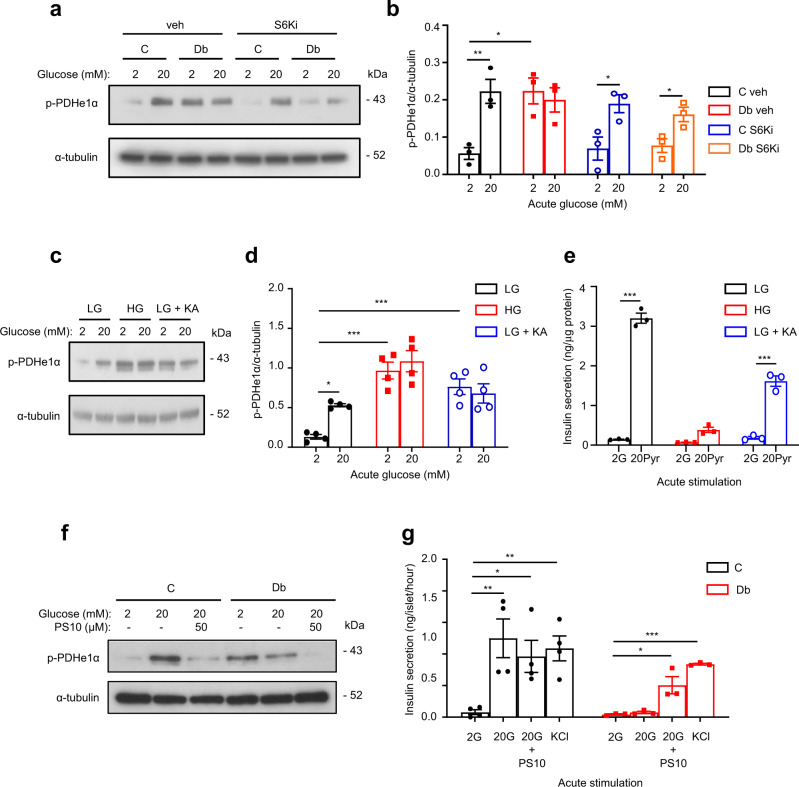
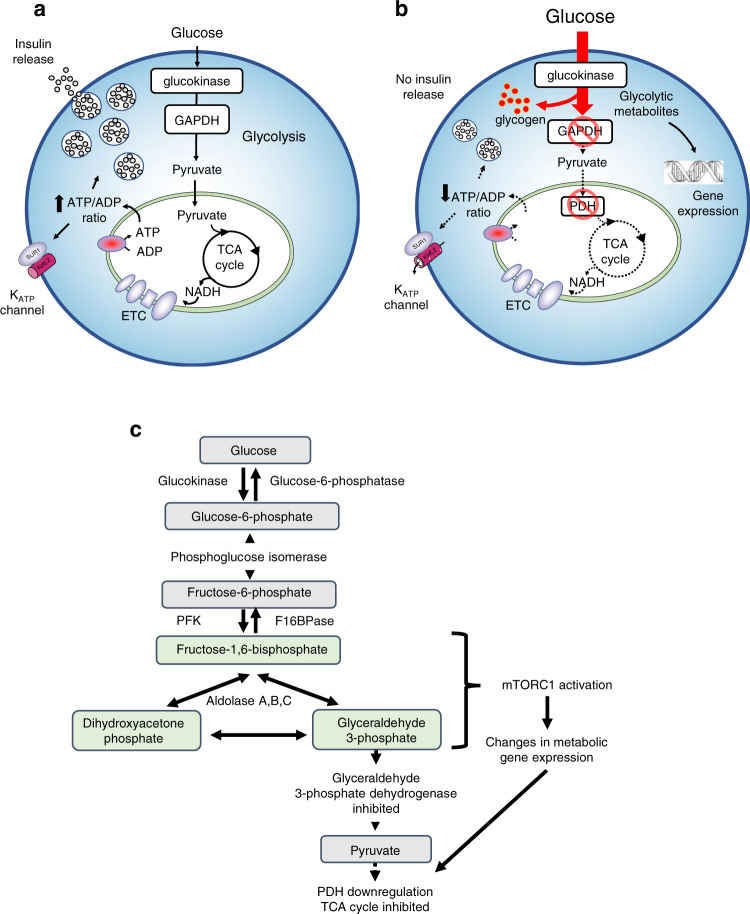
Chronic hyperglycaemia causes a dramatic decrease in mitochondrial metabolism and insulin content in pancreatic β-cells. This underlies the progressive decline in β-cell function in diabetes. However, the molecular mechanisms by which hyperglycaemia produces these effects remain unresolved. Using isolated islets and INS-1 cells, we show here that one or more glycolytic metabolites downstream of phosphofructokinase and upstream of GAPDH mediates the effects of chronic hyperglycemia. This metabolite stimulates marked upregulation of mTORC1 and concomitant downregulation of AMPK. Increased mTORC1 activity causes inhibition of pyruvate dehydrogenase which reduces pyruvate entry into the tricarboxylic acid cycle and partially accounts for the hyperglycaemia-induced reduction in oxidative phosphorylation and insulin secretion. In addition, hyperglycaemia (or diabetes) dramatically inhibits GAPDH activity, thereby impairing glucose metabolism. Our data also reveal that restricting glucose metabolism during hyperglycaemia prevents these changes and thus may be of therapeutic benefit. In summary, we have identified a pathway by which chronic hyperglycaemia reduces β-cell function.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
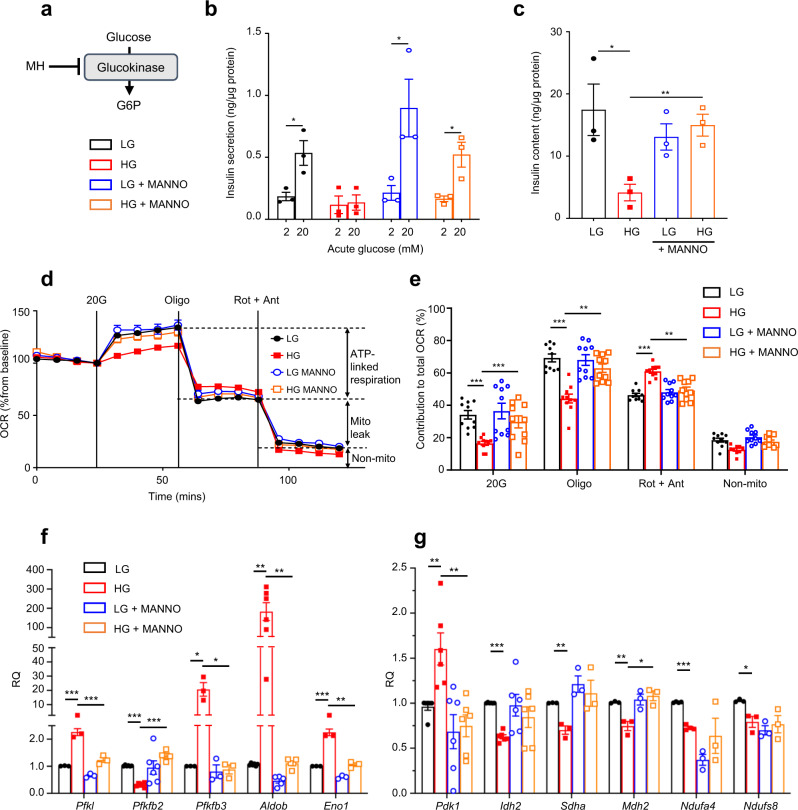
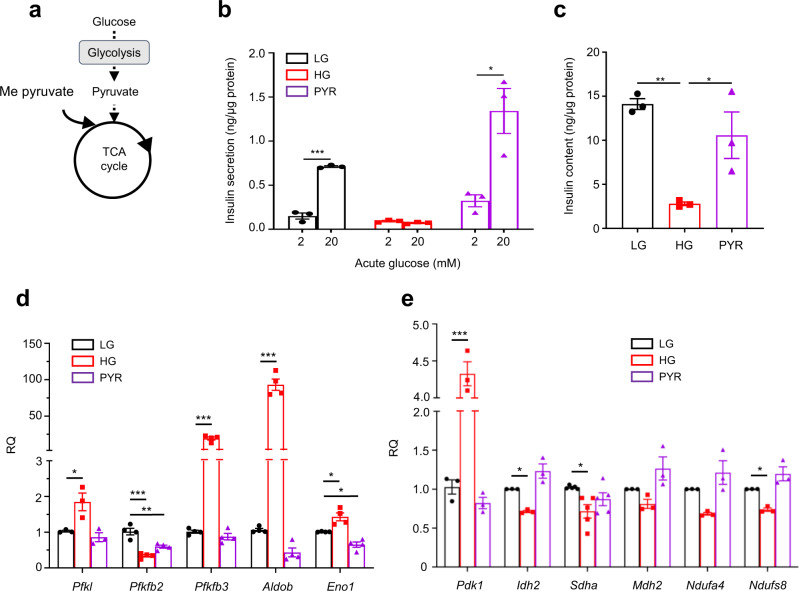
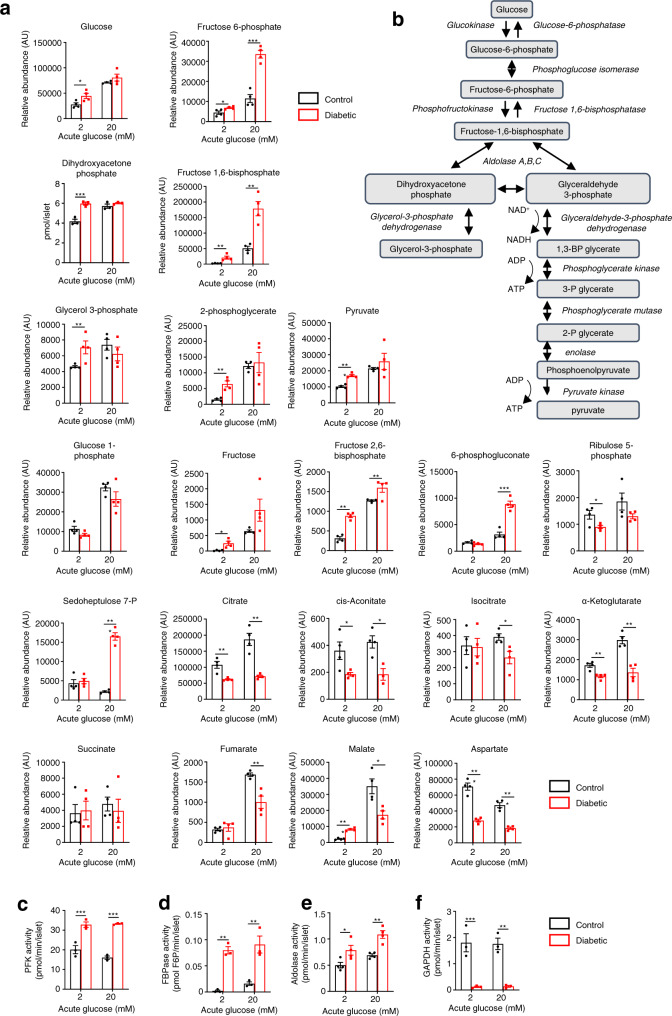
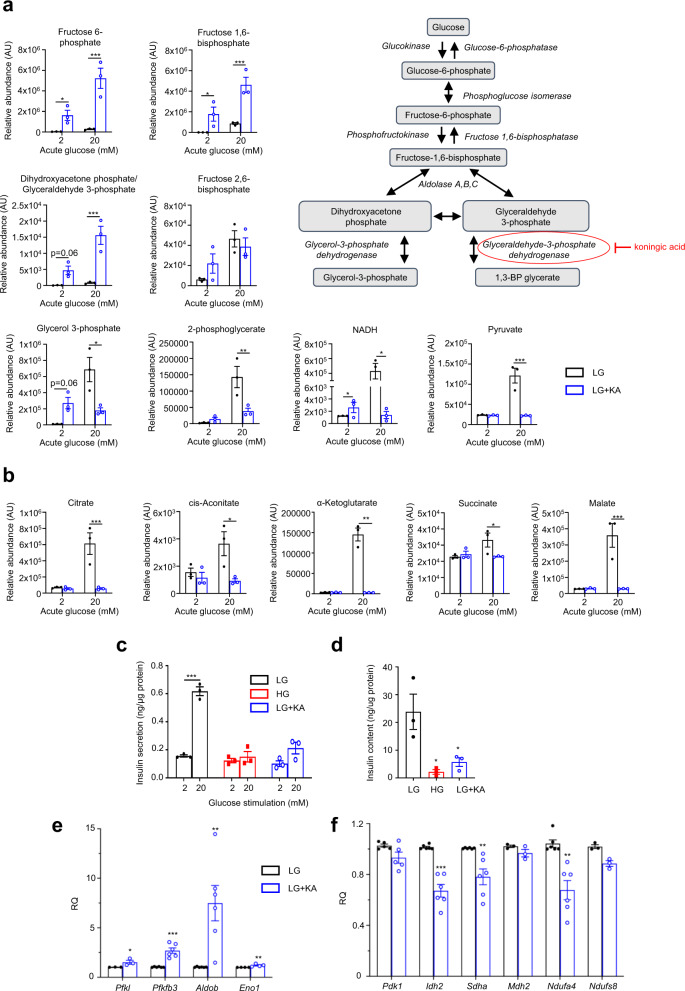
References
-
- UK Prospective Diabetes Study Group. UK Prospective Diabetes Study 16: overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes44, 1249–1258 (1995). - PubMed
-
- Marselli, L. et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia57, 362–365 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
