

The 3D enhancer network of the developing T cell genome is shaped by SATB1
- PMID: 36376298
- PMCID: PMC9663569
- DOI: 10.1038/s41467-022-34345-y
The 3D enhancer network of the developing T cell genome is shaped by SATB1
Abstract
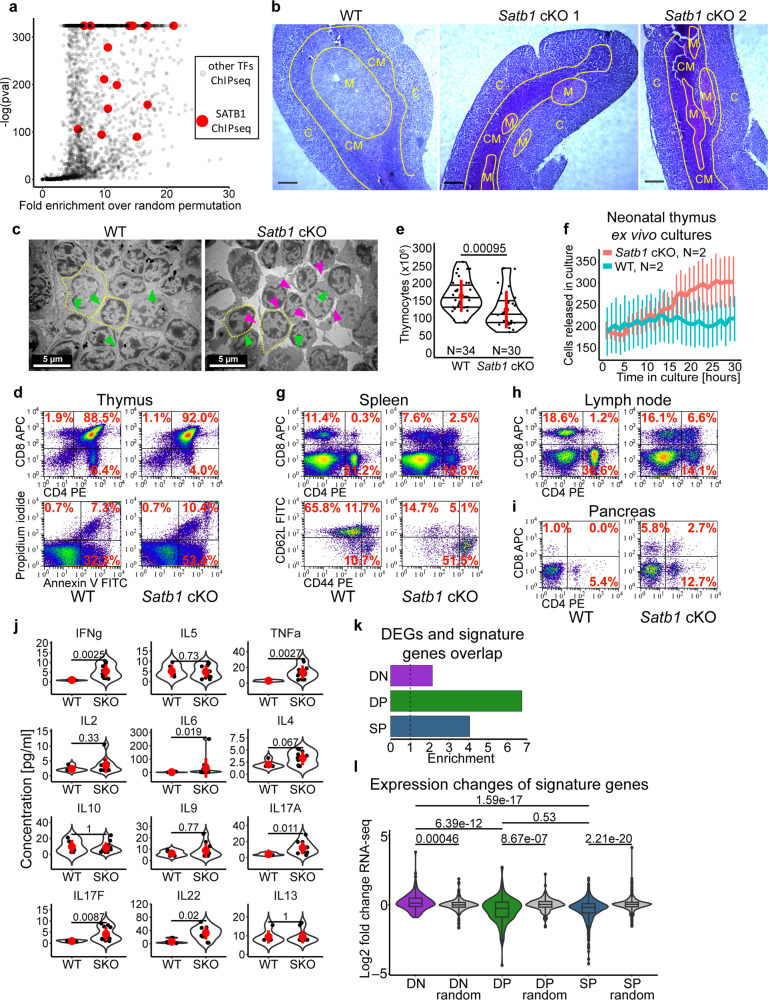
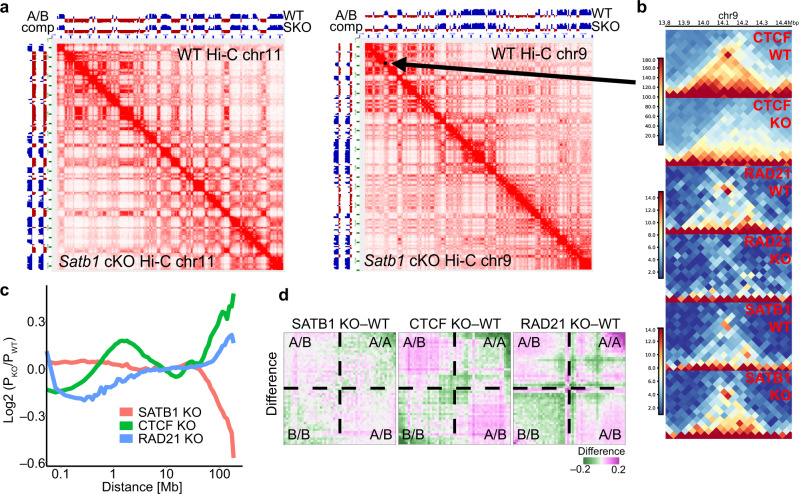
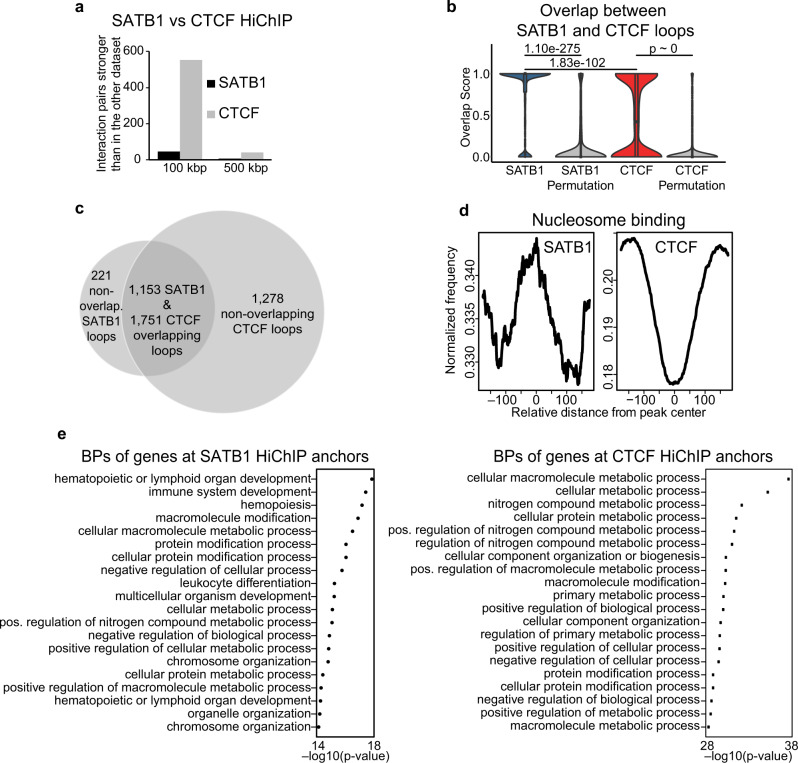
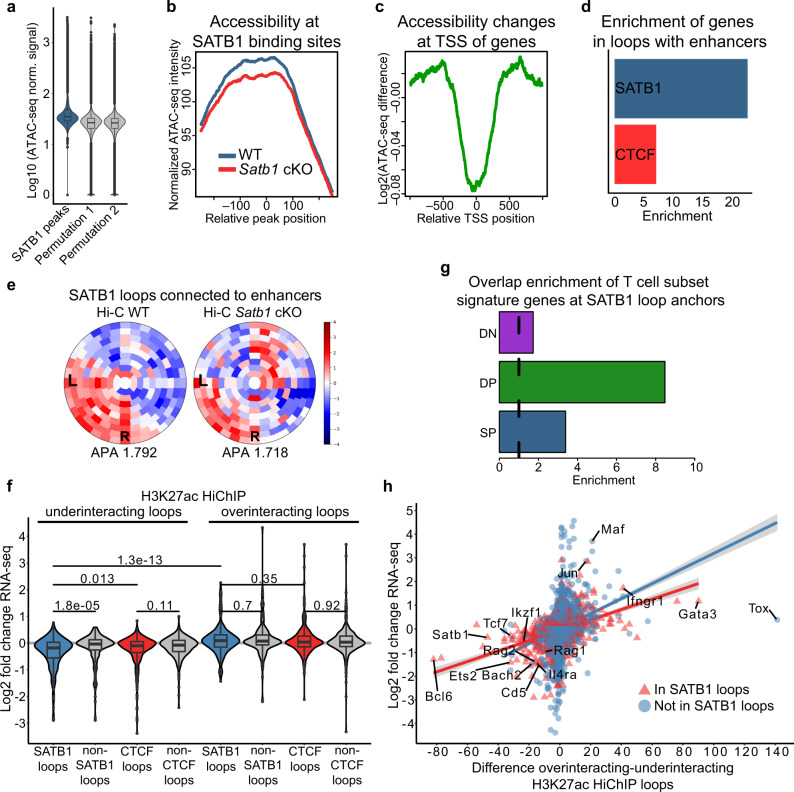
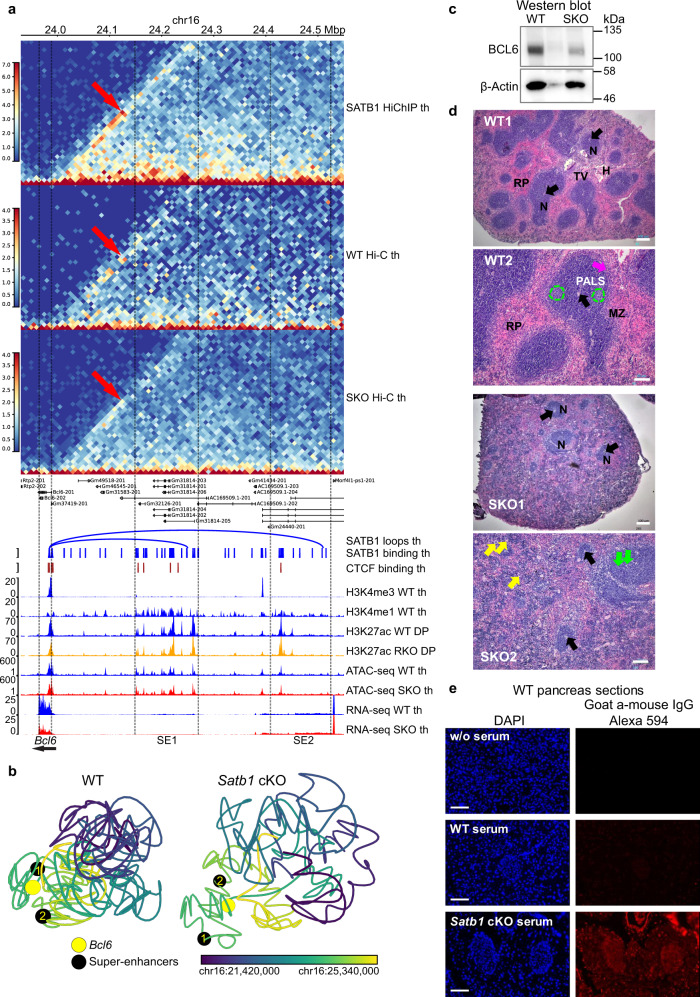
Mechanisms of tissue-specific gene expression regulation via 3D genome organization are poorly understood. Here we uncover the regulatory chromatin network of developing T cells and identify SATB1, a tissue-specific genome organizer, enriched at the anchors of promoter-enhancer loops. We have generated a T-cell specific Satb1 conditional knockout mouse which allows us to infer the molecular mechanisms responsible for the deregulation of its immune system. H3K27ac HiChIP and Hi-C experiments indicate that SATB1-dependent promoter-enhancer loops regulate expression of master regulator genes (such as Bcl6), the T cell receptor locus and adhesion molecule genes, collectively being critical for cell lineage specification and immune system homeostasis. SATB1-dependent regulatory chromatin loops represent a more refined layer of genome organization built upon a high-order scaffold provided by CTCF and other factors. Overall, our findings unravel the function of a tissue-specific factor that controls transcription programs, via spatial chromatin arrangements complementary to the chromatin structure imposed by ubiquitously expressed genome organizers.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
