

Beyond PI3Ks: targeting phosphoinositide kinases in disease
- PMID: 36376561
- PMCID: PMC9663198
- DOI: 10.1038/s41573-022-00582-5
Beyond PI3Ks: targeting phosphoinositide kinases in disease
Abstract
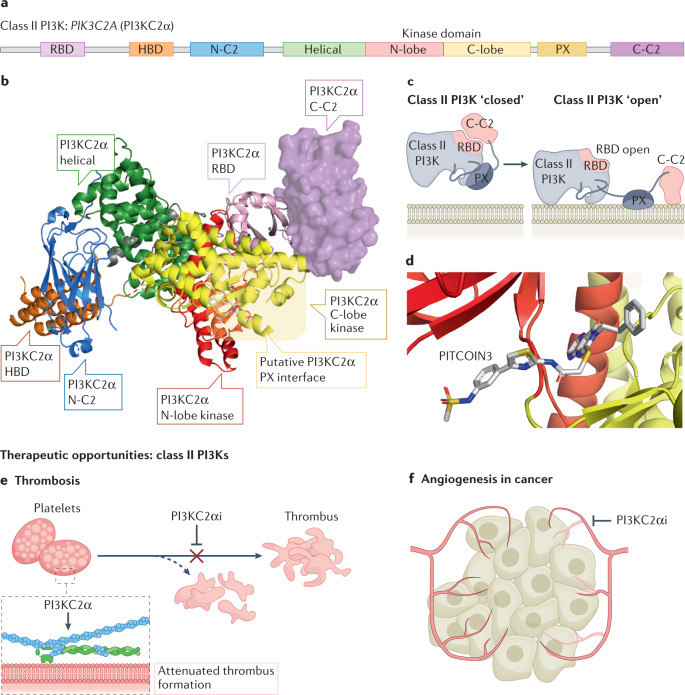
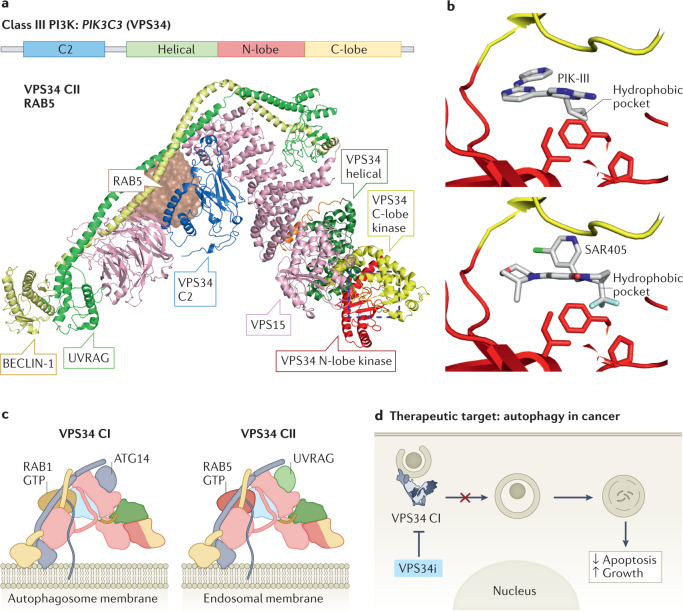
Lipid phosphoinositides are master regulators of almost all aspects of a cell's life and death and are generated by the tightly regulated activity of phosphoinositide kinases. Although extensive efforts have focused on drugging class I phosphoinositide 3-kinases (PI3Ks), recent years have revealed opportunities for targeting almost all phosphoinositide kinases in human diseases, including cancer, immunodeficiencies, viral infection and neurodegenerative disease. This has led to widespread efforts in the clinical development of potent and selective inhibitors of phosphoinositide kinases. This Review summarizes our current understanding of the molecular basis for the involvement of phosphoinositide kinases in disease and assesses the preclinical and clinical development of phosphoinositide kinase inhibitors.
© 2022. Springer Nature Limited.
Conflict of interest statement
J.E.B. reports personal fees from Scorpion Therapeutics and Olema Oncology; and research grants from Novartis. J.T., B.M.E. and G.R.V.H. declare no competing interests.
Figures
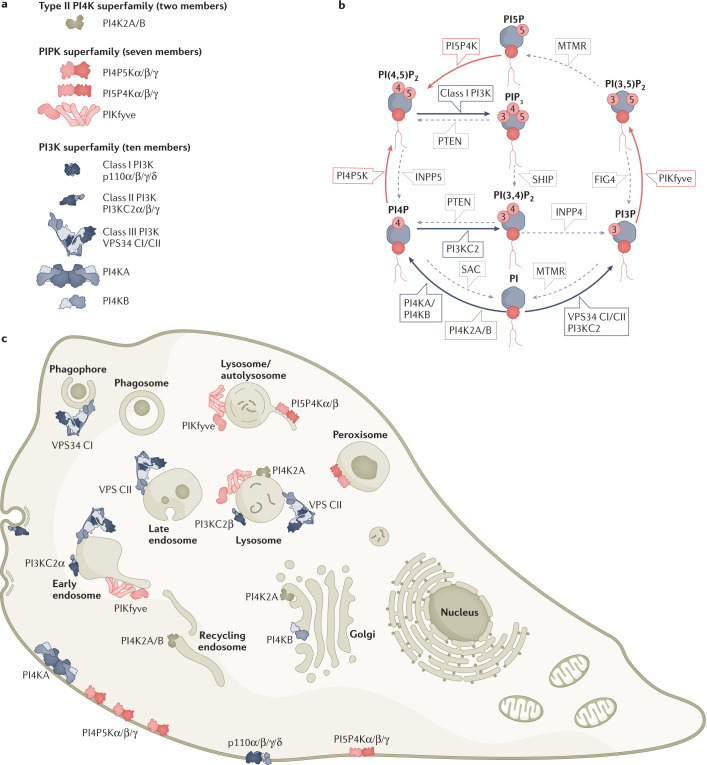
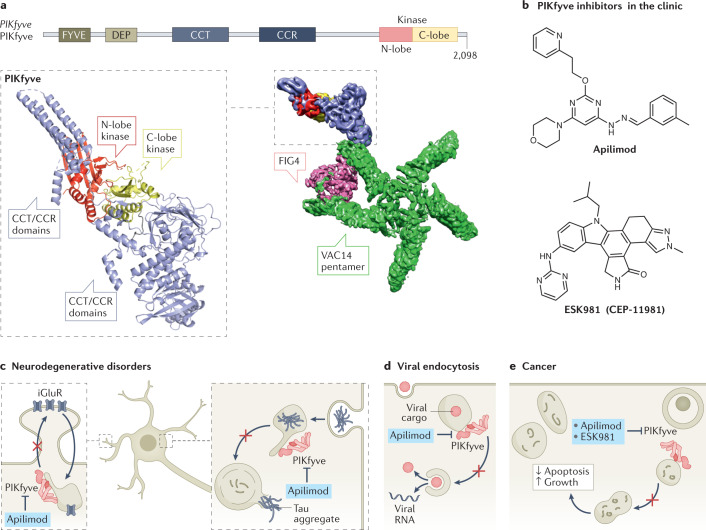
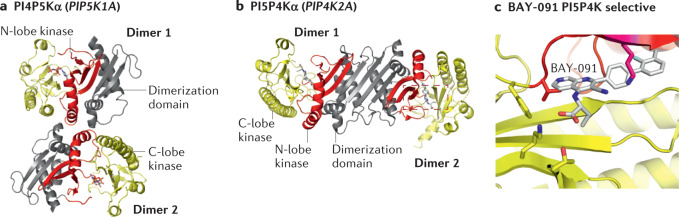
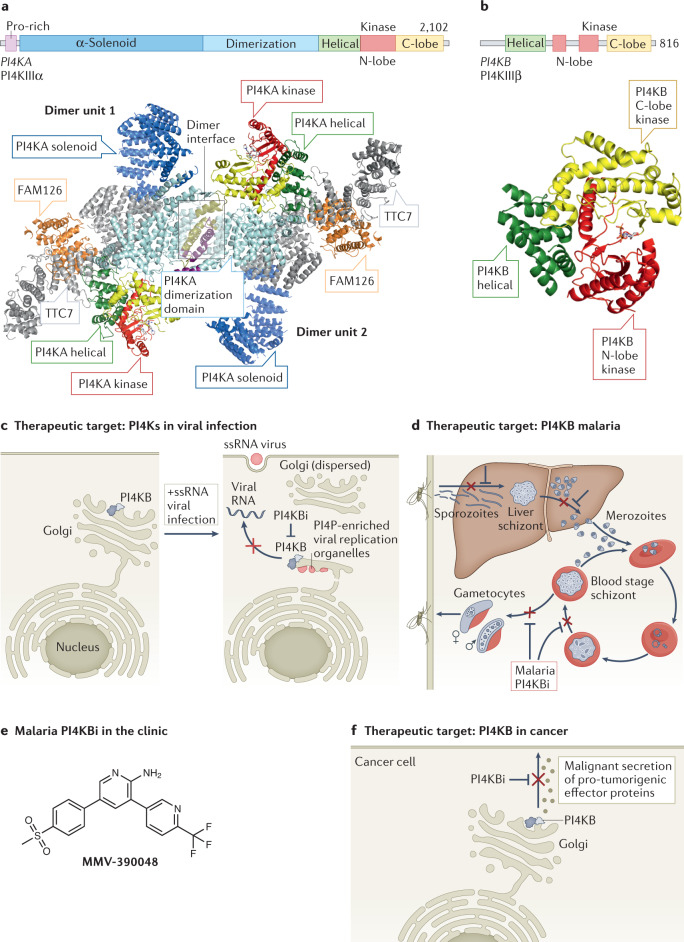
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
