

SCIP: software for efficient clinical interpretation of copy number variants detected by whole-genome sequencing
- PMID: 36376761
- PMCID: PMC9918589
- DOI: 10.1007/s00439-022-02494-1
SCIP: software for efficient clinical interpretation of copy number variants detected by whole-genome sequencing
Abstract
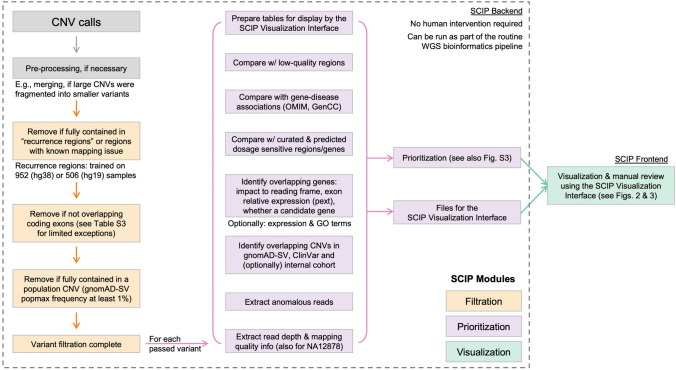
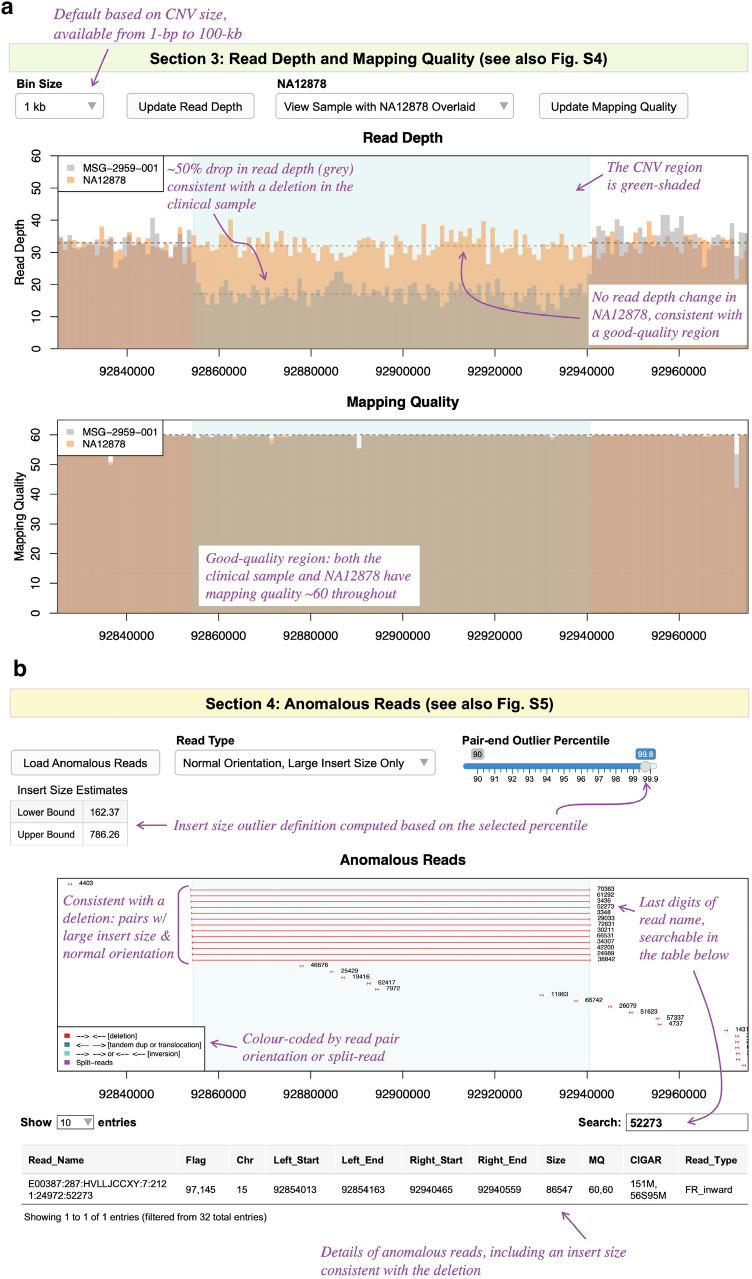
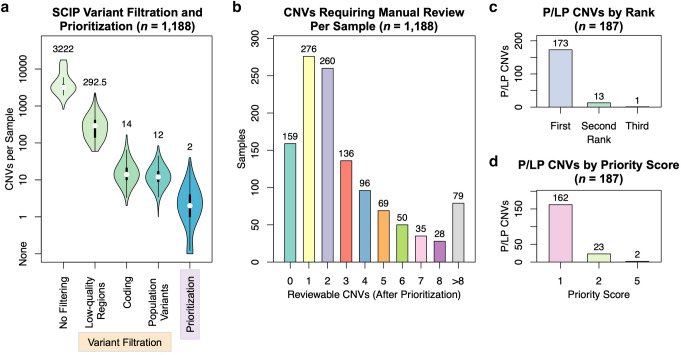
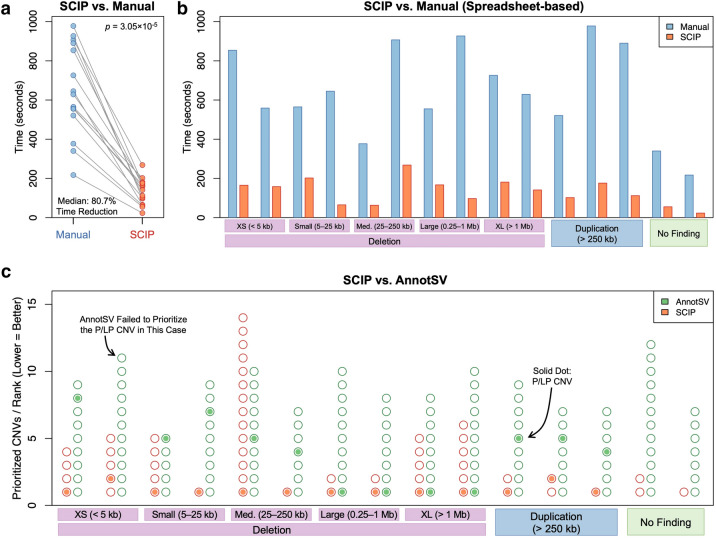
Copy number variants (CNVs) represent major etiologic factors in rare genetic diseases. Current clinical CNV interpretation workflows require extensive back-and-forth with multiple tools and databases. This increases complexity and time burden, potentially resulting in missed genetic diagnoses. We present the Suite for CNV Interpretation and Prioritization (SCIP), a software package for the clinical interpretation of CNVs detected by whole-genome sequencing (WGS). The SCIP Visualization Module near-instantaneously displays all information necessary for CNV interpretation (variant quality, population frequency, inheritance pattern, and clinical relevance) on a single page-supported by modules providing variant filtration and prioritization. SCIP was comprehensively evaluated using WGS data from 1027 families with congenital cardiac disease and/or autism spectrum disorder, containing 187 pathogenic or likely pathogenic (P/LP) CNVs identified in previous curations. SCIP was efficient in filtration and prioritization: a median of just two CNVs per case were selected for review, yet it captured all P/LP findings (92.5% of which ranked 1st). SCIP was also able to identify one pathogenic CNV previously missed. SCIP was benchmarked against AnnotSV and a spreadsheet-based manual workflow and performed superiorly than both. In conclusion, SCIP is a novel software package for efficient clinical CNV interpretation, substantially faster and more accurate than previous tools (available at https://github.com/qd29/SCIP , a video tutorial series is available at https://bit.ly/SCIPVideos ).
© 2022. The Author(s).
Conflict of interest statement
None declared.
Figures



References
-
- Austin-Tse CA, Jobanputra V, Perry DL, Bick D, Taft RJ, Venner E, Gibbs RA, Young T, Barnett S, Belmont JW, Boczek N, Chowdhury S, Ellsworth KA, Guha S, Kulkarni S, Marcou C, Meng L, Murdock DR, Rehman AU, Spiteri E, Thomas-Wilson A, Kearney HM, Rehm HL. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom Med. 2022;7:27. doi: 10.1038/s41525-022-00295-z. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
