

Clinical characterizations of three adults with genetically confirmed spinal muscular atrophy: a case series
- PMID: 36376972
- PMCID: PMC9664805
- DOI: 10.1186/s13256-022-03633-y
Clinical characterizations of three adults with genetically confirmed spinal muscular atrophy: a case series
Abstract
Background: Spinal muscular atrophy is a recessively inherited autosomal neuromuscular disorder, with characteristic progressive muscle weakness. Most spinal muscular atrophy cases clinically manifest during infancy or childhood, although it may first manifest in adulthood. Although spinal muscular atrophy has come to the era of newborn screening and promising treatments, genetically confirmed spinal muscular atrophy patients are still rare in third world countries, including Indonesia.
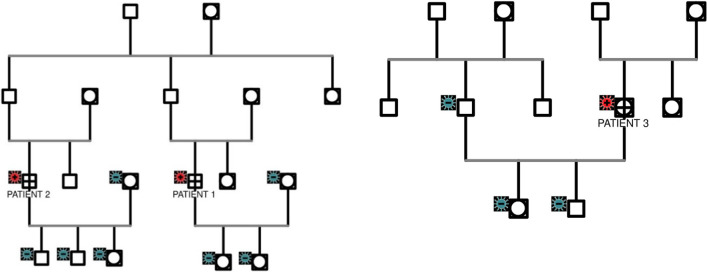
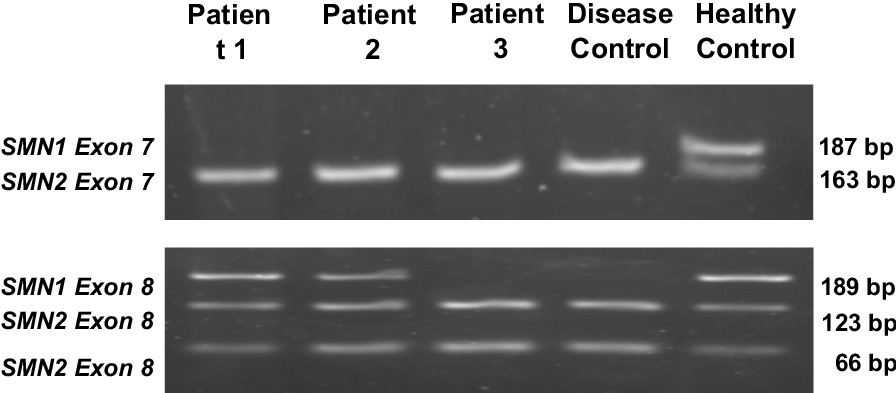
Case presentations: We presented three Indonesian patients with spinal muscular atrophy genetically confirmed during adulthood. The first case was a 40-year-old male who presented with weakness in his lower limbs that started when he was 9 years old. At the age of 16 years, he could no longer walk and started using a wheelchair. He first came to our clinic at the age of 38 years, and was diagnosed with spinal muscular atrophy 2 years later. The second patient was a 58-year-old male who presented with lower limb weakness since he was 12 years old. Owing to the geographical distance and financial problems, he was referred to our clinic at the age of 56 years, when he already used a walker to walk. Lastly, the third patient was a 28-year-old woman, who was in the first semester of her second pregnancy, and who presented with slowly progressing lower limb weakness. Her limb weakness began at the age of 8 years, and slowly progressed until she became dependent on her wheelchair 8 years later until now. She had successfully given birth to a healthy daughter 3 years before her first visit to our clinic. All three patients were diagnosed with neuromuscular disorder diseases, with the differential diagnoses of Duchenne muscular dystrophy, spinal muscular atrophy, and Becker muscular dystrophy. These patients were finally confirmed to have spinal muscular atrophy due to SMN1 deletion by polymerase chain reaction and restriction fragment length polymorphism.
Conclusions: Many genetic diseases are often neglected in developing countries owing to the difficulty in diagnosis and unavailable treatment. Our case series focused on the disease courses, diagnosis difficulties, and clinical presentations of three patients that finally lead to diagnoses of spinal muscular atrophy.
Keywords: Clinical characterization; Diagnosis; Indonesia; Spinal muscular atrophy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
