

SARS-CoV-2 Genomic Diversity in Households Highlights the Challenges of Sequence-Based Transmission Inference
- PMID: 36377913
- PMCID: PMC9769559
- DOI: 10.1128/msphere.00400-22
SARS-CoV-2 Genomic Diversity in Households Highlights the Challenges of Sequence-Based Transmission Inference
Abstract
The reliability of sequence-based inference of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission is not clear. Sequence data from infections among household members can define the expected genomic diversity of a virus along a defined transmission chain. SARS-CoV-2 cases were identified prospectively among 2,369 participants in 706 households. Specimens with a reverse transcription-PCR cycle threshold of ≤30 underwent whole-genome sequencing. Intrahost single-nucleotide variants (iSNV) were identified at a ≥5% frequency. Phylogenetic trees were used to evaluate the relationship of household and community sequences. There were 178 SARS-CoV-2 cases in 706 households. Among 147 specimens sequenced, 106 yielded a whole-genome consensus with coverage suitable for identifying iSNV. Twenty-six households had sequences from multiple cases within 14 days. Consensus sequences were indistinguishable among cases in 15 households, while 11 had ≥1 consensus sequence that differed by 1 to 2 mutations. Sequences from households and the community were often interspersed on phylogenetic trees. Identification of iSNV improved inference in 2 of 15 households with indistinguishable consensus sequences and in 6 of 11 with distinct ones. In multiple-infection households, whole-genome consensus sequences differed by 0 to 1 mutations. Identification of shared iSNV occasionally resolved linkage, but the low genomic diversity of SARS-CoV-2 limits the utility of "sequence-only" transmission inference. IMPORTANCE We performed whole-genome sequencing of SARS-CoV-2 from prospectively identified cases in three longitudinal household cohorts. In a majority of multi-infection households, SARS-CoV-2 consensus sequences were indistinguishable, and they differed by 1 to 2 mutations in the rest. Importantly, even with modest genomic surveillance of the community (3 to 5% of cases sequenced), it was not uncommon to find community sequences interspersed with household sequences on phylogenetic trees. Identification of shared minority variants only occasionally resolved these ambiguities in transmission linkage. Overall, the low genomic diversity of SARS-CoV-2 limits the utility of "sequence-only" transmission inference. Our work highlights the need to carefully consider both epidemiologic linkage and sequence data to define transmission chains in households, hospitals, and other transmission settings.
Keywords: SARS-CoV-2; genomic epidemiology; household; transmission.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
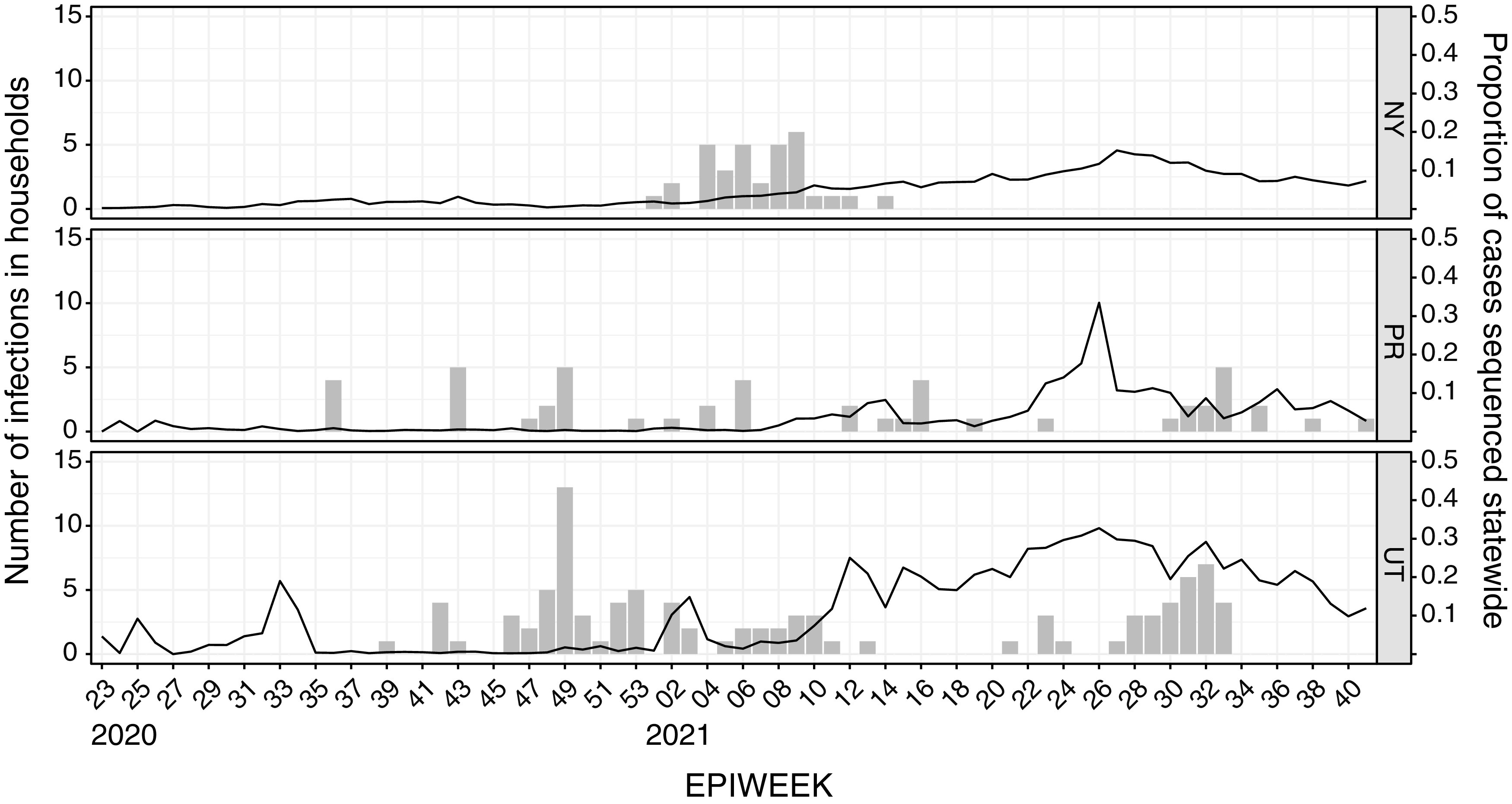
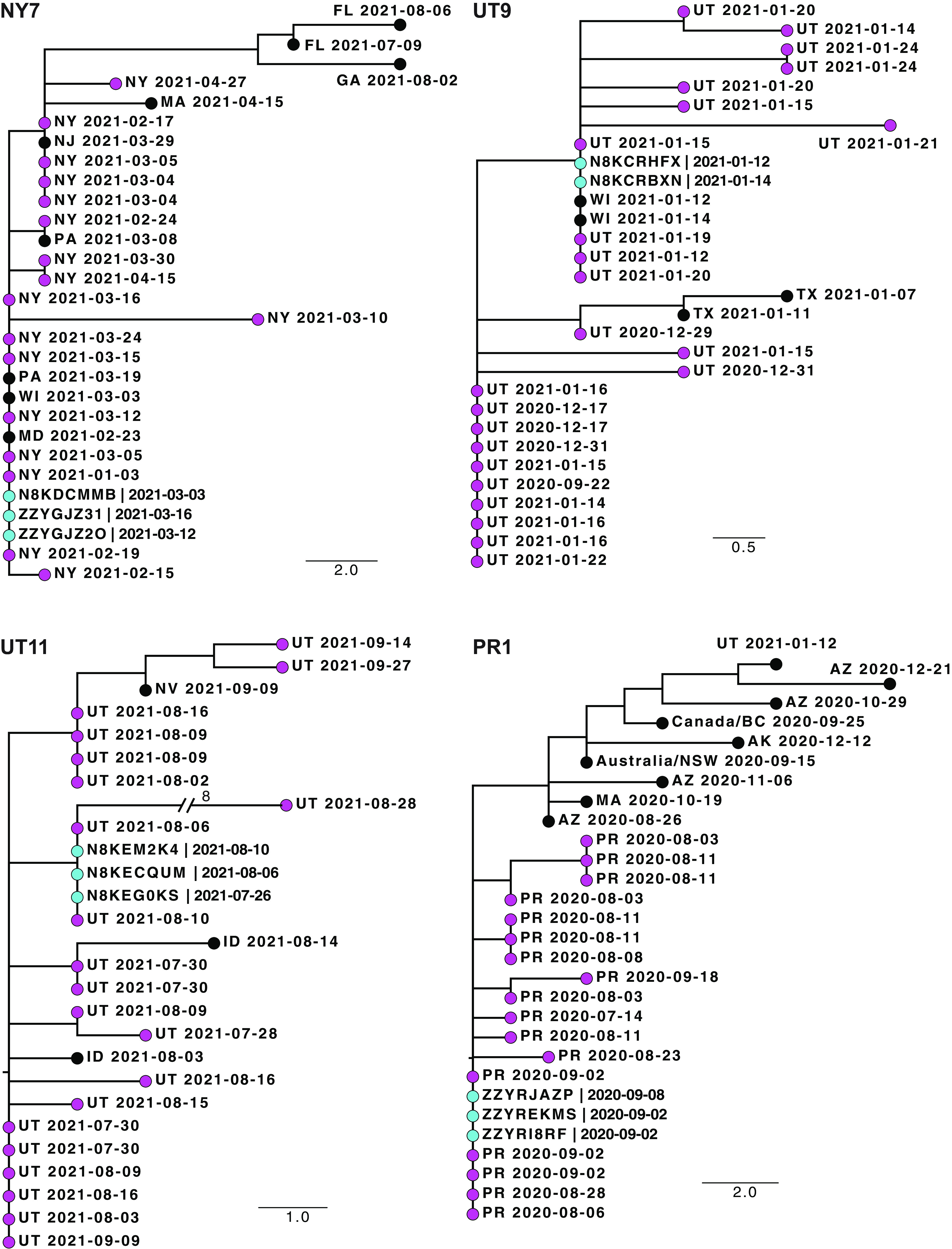
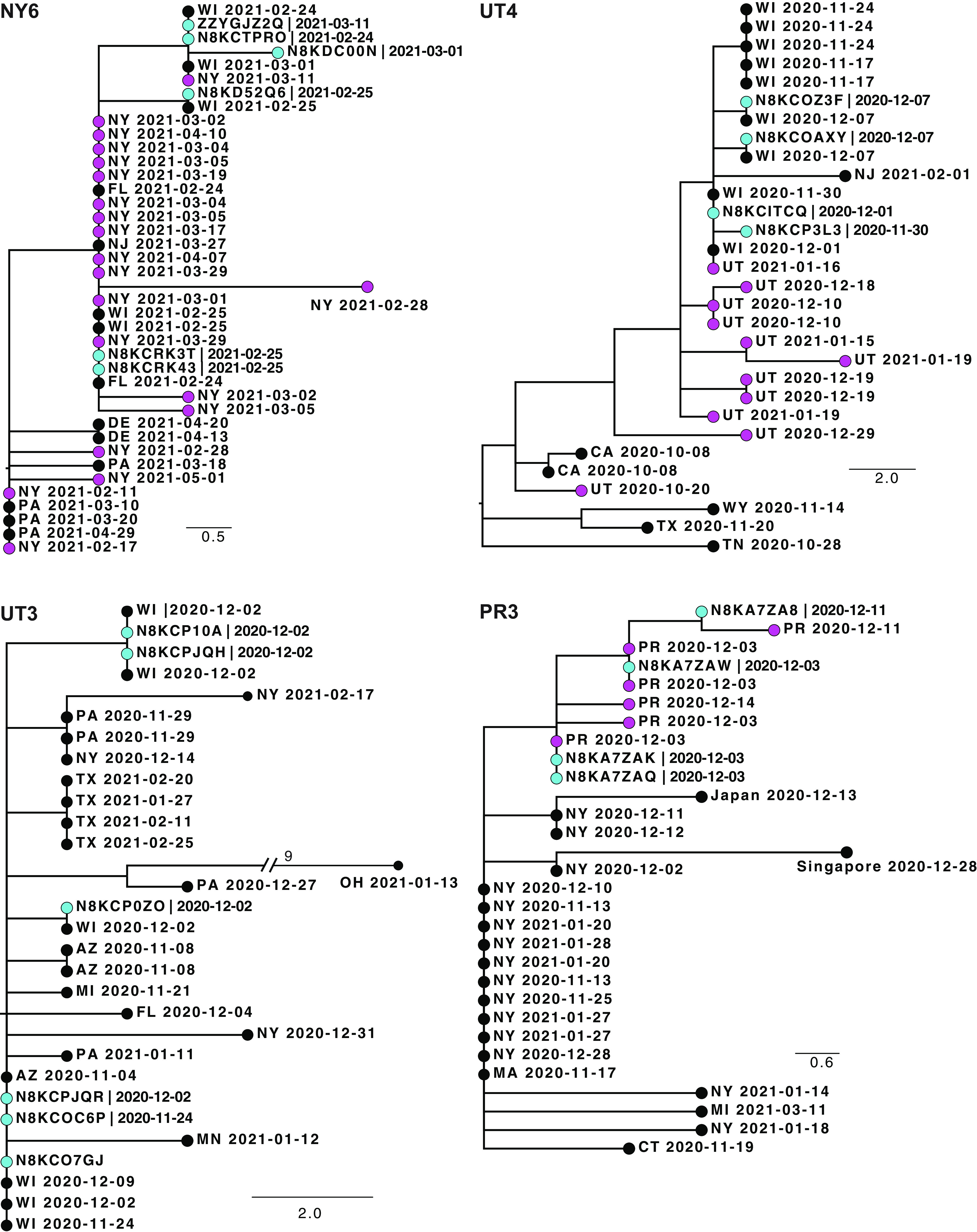
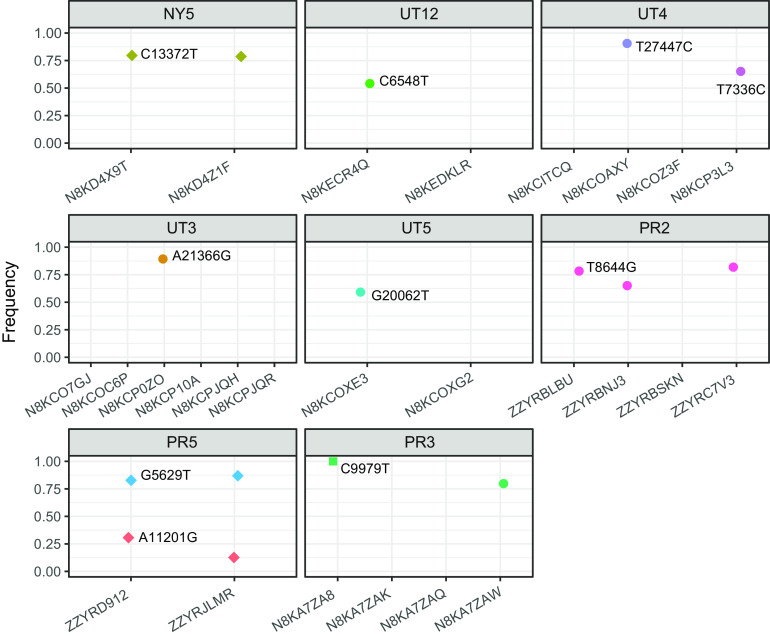
Comment in
-
Low Viral Diversity Limits the Effectiveness of Sequence-Based Transmission Inference for SARS-CoV-2.mSphere. 2023 Feb 21;8(1):e0054422. doi: 10.1128/msphere.00544-22. Epub 2023 Jan 25. mSphere. 2023. PMID: 36695609 Free PMC article.
References
-
- Lemieux JE, Siddle KJ, Shaw BM, Loreth C, Schaffner SF, Gladden-Young A, Adams G, Fink T, Tomkins-Tinch CH, Krasilnikova LA, DeRuff KC, Rudy M, Bauer MR, Lagerborg KA, Normandin E, Chapman SB, Reilly SK, Anahtar MN, Lin AE, Carter A, Myhrvold C, Kemball ME, Chaluvadi S, Cusick C, Flowers K, Neumann A, Cerrato F, Farhat M, Slater D, Harris JB, Branda JA, Hooper D, Gaeta JM, Baggett TP, O’Connell J, Gnirke A, Lieberman TD, Philippakis A, Burns M, Brown CM, Luban J, Ryan ET, Turbett SE, LaRocque RC, Hanage WP, Gallagher GR, Madoff LC, Smole S, Pierce VM, Rosenberg E, et al. 2021. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science 371:eabe3261. doi: 10.1126/science.abe3261. - DOI - PMC - PubMed
-
- Siddle KJ, Krasilnikova LA, Moreno GK, Schaffner SF, Vostok J, Fitzgerald NA, Lemieux JE, Barkas N, Loreth C, Specht I, Tomkins-Tinch CH, Paull JS, Schaeffer B, Taylor BP, Loftness B, Johnson H, Schubert PL, Shephard HM, Doucette M, Fink T, Lang AS, Baez S, Beauchamp J, Hennigan S, Buzby E, Ash S, Brown J, Clancy S, Cofsky S, Gagne L, Hall J, Harrington R, Gionet GL, DeRuff KC, Vodzak ME, Adams GC, Dobbins ST, Slack SD, Reilly SK, Anderson LM, Cipicchio MC, DeFelice MT, Grimsby JL, Anderson SE, Blumenstiel BS, Meldrim JC, Rooke HM, Vicente G, Smith NL, Messer KS, et al. 2022. Transmission from vaccinated individuals in a large SARS-CoV-2 Delta variant outbreak. Cell 185:485–492.e10. doi: 10.1016/j.cell.2021.12.027. - DOI - PMC - PubMed
-
- Zeller M, Gangavarapu K, Anderson C, Smither AR, Vanchiere JA, Rose R, Snyder DJ, Dudas G, Watts A, Matteson NL, Robles-Sikisaka R, Marshall M, Feehan AK, Sabino-Santos G, Bell-Kareem AR, Hughes LD, Alkuzweny M, Snarski P, Garcia-Diaz J, Scott RS, Melnik LI, Klitting R, McGraw M, Belda-Ferre P, DeHoff P, Sathe S, Marotz C, Grubaugh ND, Nolan DJ, Drouin AC, Genemaras KJ, Chao K, Topol S, Spencer E, Nicholson L, Aigner S, Yeo GW, Farnaes L, Hobbs CA, Laurent LC, Knight R, Hodcroft EB, Khan K, Fusco DN, Cooper VS, Lemey P, Gardner L, Lamers SL, Kamil JP, Garry RF, et al. 2021. Emergence of an early SARS-CoV-2 epidemic in the United States. Cell 184:4939–4952.e15. doi: 10.1016/j.cell.2021.07.030. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
