

Multiple-low-dose therapy: effective killing of high-grade serous ovarian cancer cells with ATR and CHK1 inhibitors
- PMID: 36381271
- PMCID: PMC9653014
- DOI: 10.1093/narcan/zcac036
Multiple-low-dose therapy: effective killing of high-grade serous ovarian cancer cells with ATR and CHK1 inhibitors
Abstract
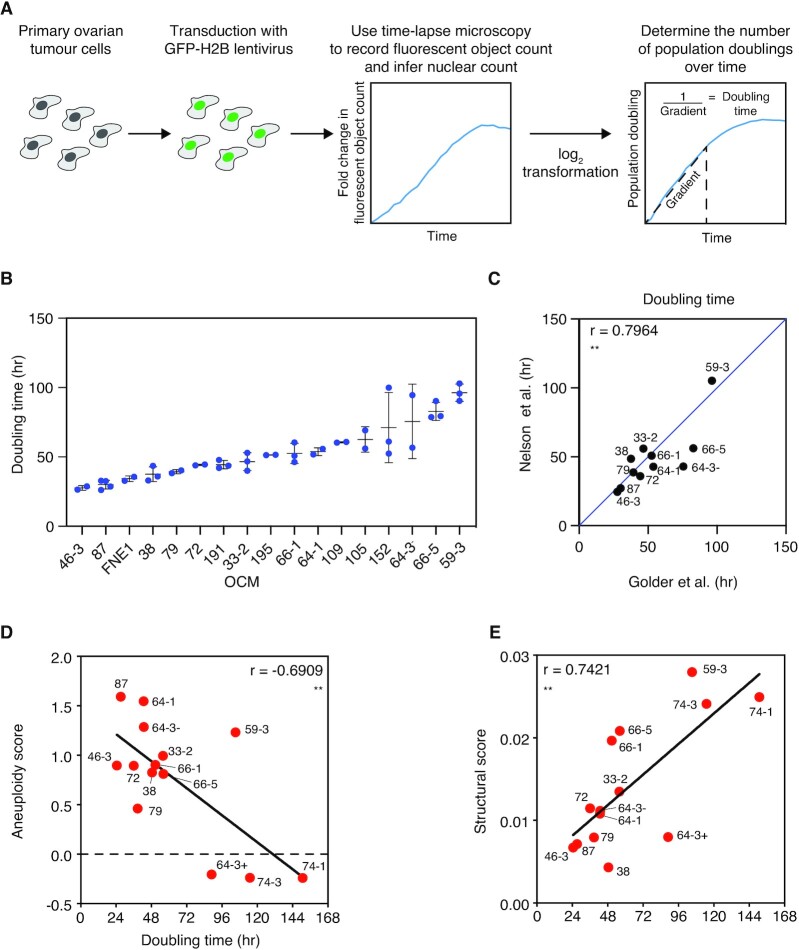
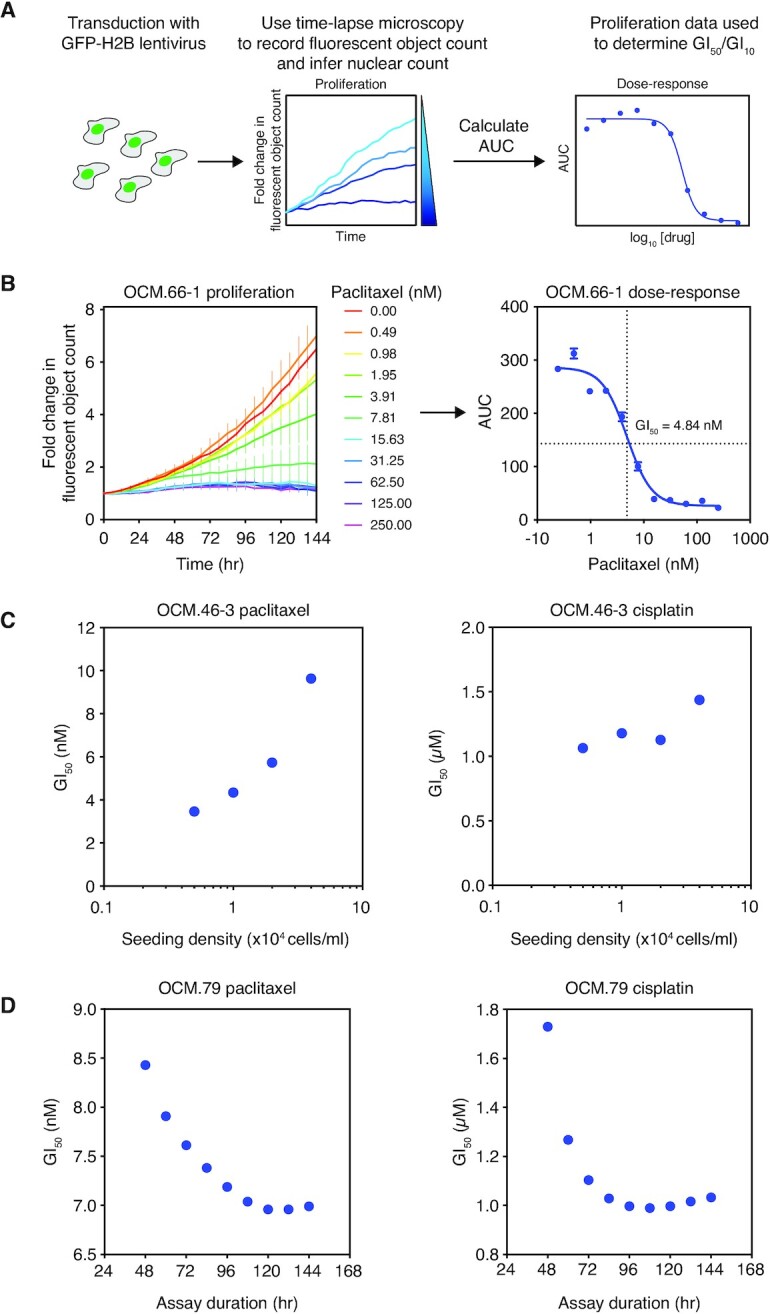
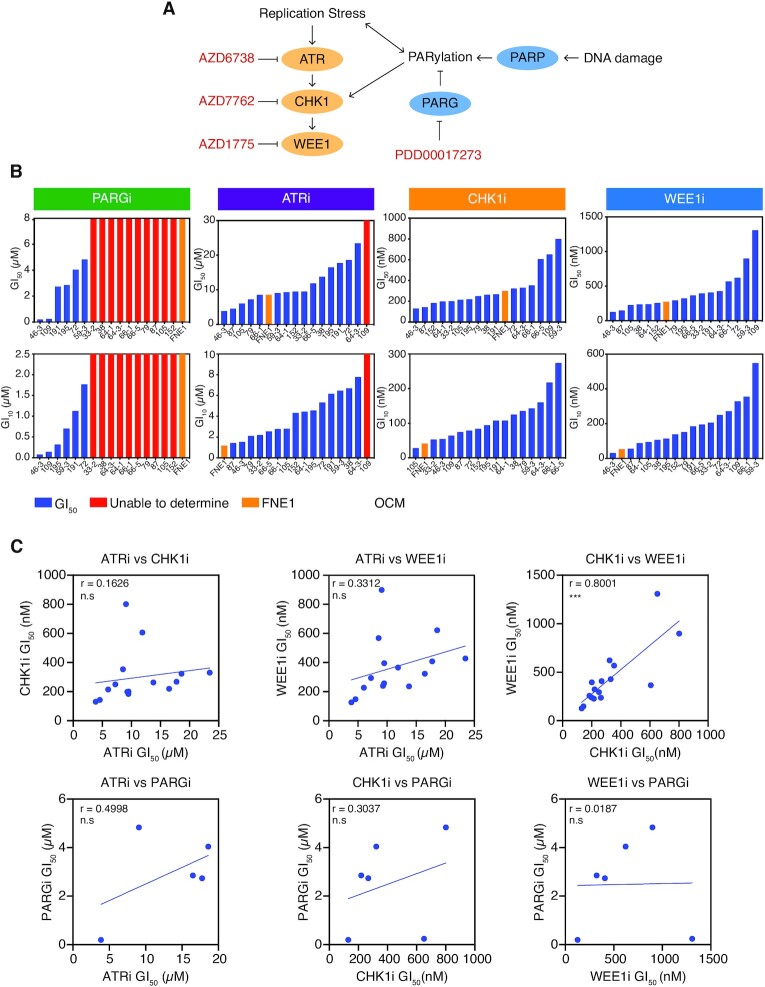
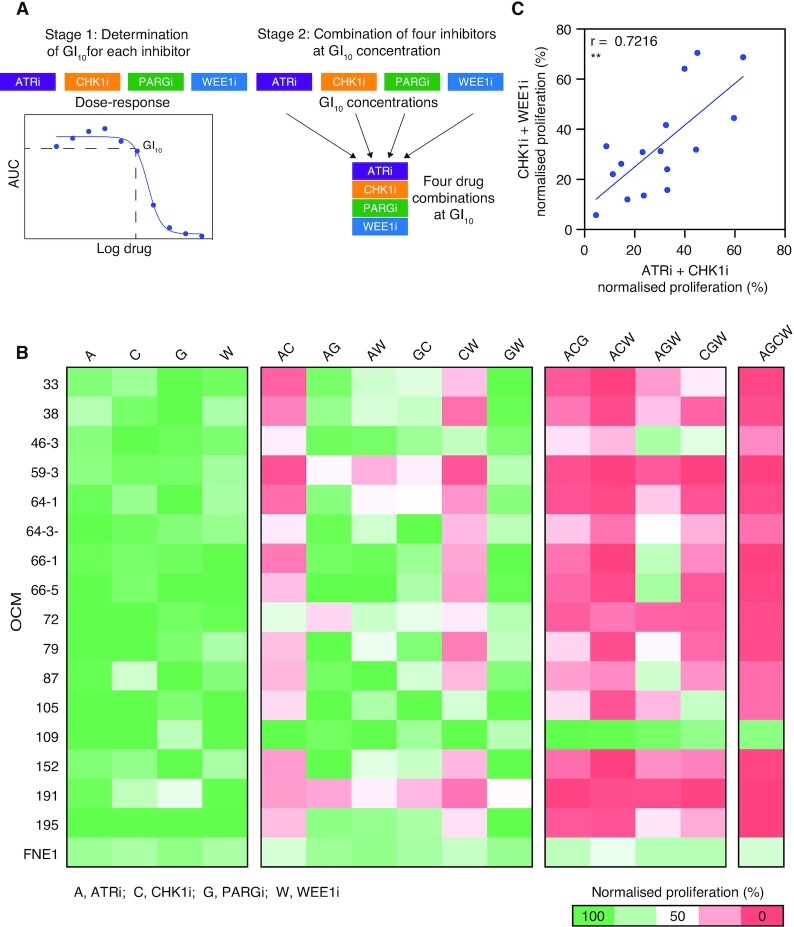
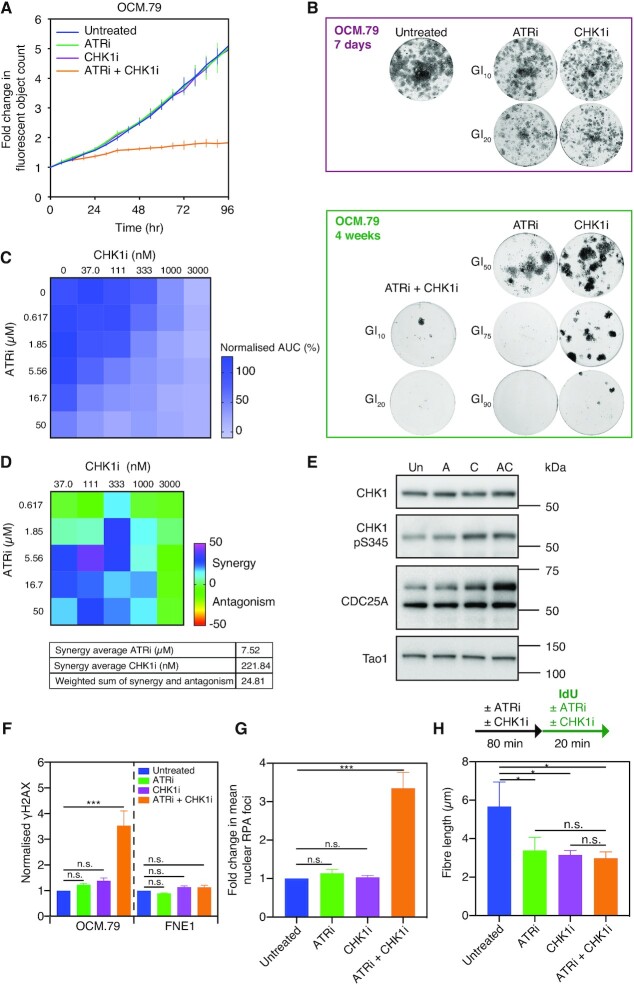
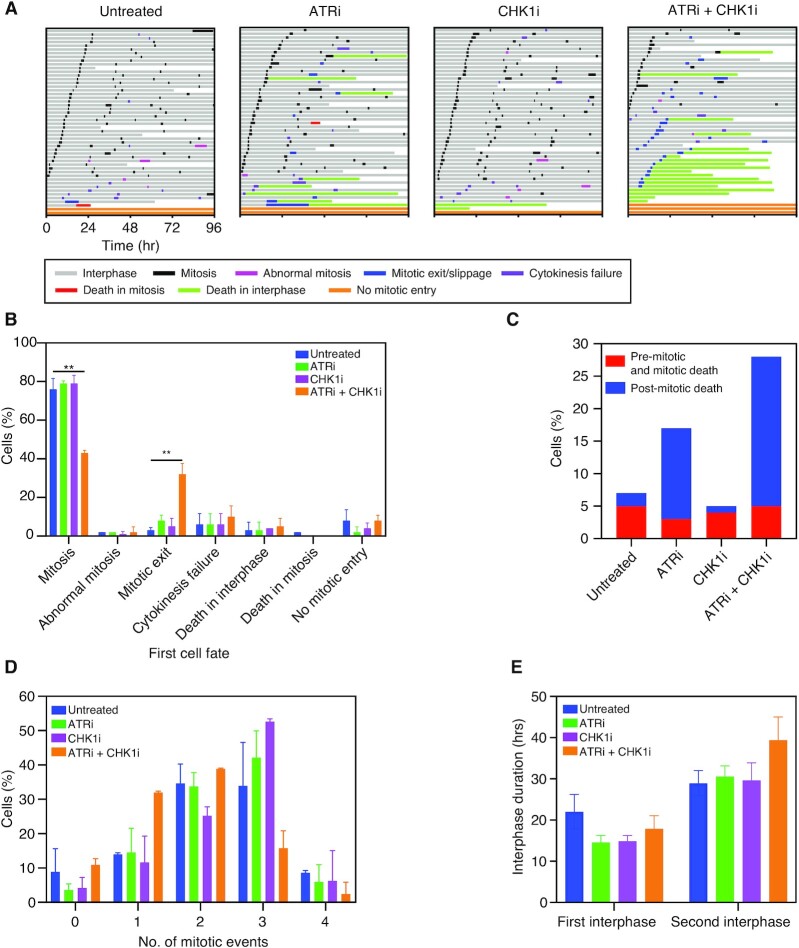
High-grade serous ovarian cancer (HGSOC) is an aggressive disease that typically develops drug resistance, thus novel biomarker-driven strategies are required. Targeted therapy focuses on synthetic lethality-pioneered by PARP inhibition of BRCA1/2-mutant disease. Subsequently, targeting the DNA replication stress response (RSR) is of clinical interest. However, further mechanistic insight is required for biomarker discovery, requiring sensitive models that closely recapitulate HGSOC. We describe an optimized proliferation assay that we use to screen 16 patient-derived ovarian cancer models (OCMs) for response to RSR inhibitors (CHK1i, WEE1i, ATRi, PARGi). Despite genomic heterogeneity characteristic of HGSOC, measurement of OCM proliferation was reproducible and reflected intrinsic tumour-cell properties. Surprisingly, RSR targeting drugs were not interchangeable, as sensitivity to the four inhibitors was not correlated. Therefore, to overcome RSR redundancy, we screened the OCMs with all two-, three- and four-drug combinations in a multiple-low-dose strategy. We found that low-dose CHK1i-ATRi had a potent anti-proliferative effect on 15 of the 16 OCMs, and was synergistic with potential to minimise treatment resistance and toxicity. Low-dose ATRi-CHK1i induced replication catastrophe followed by mitotic exit and post-mitotic arrest or death. Therefore, this study demonstrates the potential of the living biobank of OCMs as a drug discovery platform for HGSOC.
© The Author(s) 2022. Published by Oxford University Press on behalf of NAR Cancer.
Figures
References
-
- Swain S.M., Miles D., Kim S.B., Im Y.H., Im S.A., Semiglazov V., Ciruelos E., Schneeweiss A., Loi S., Monturus E.et al.. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020; 21:519–530. - PubMed
-
- Tan A.C., Tan D.S.W.. Targeted therapies for lung cancer patients with oncogenic driver molecular alterations. J. Clin. Oncol. 2022; Jco2101626. - PubMed
-
- Hauschild A., Ascierto P.A., Schadendorf D., Grob J.J., Ribas A., Kiecker F., Dutriaux C., Demidov L.V., Lebbé C., Rutkowski P.et al.. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib monotherapy: analysis from phase 2 and 3 clinical trials. Eur. J. Cancer. 2020; 125:114–120. - PMC - PubMed
-
- Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T.. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
