

Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks [Article v0.1]
- PMID: 36382113
- PMCID: PMC9662604
- DOI: 10.33011/livecoms.4.1.1497
Best practices for constructing, preparing, and evaluating protein-ligand binding affinity benchmarks [Article v0.1]
Abstract
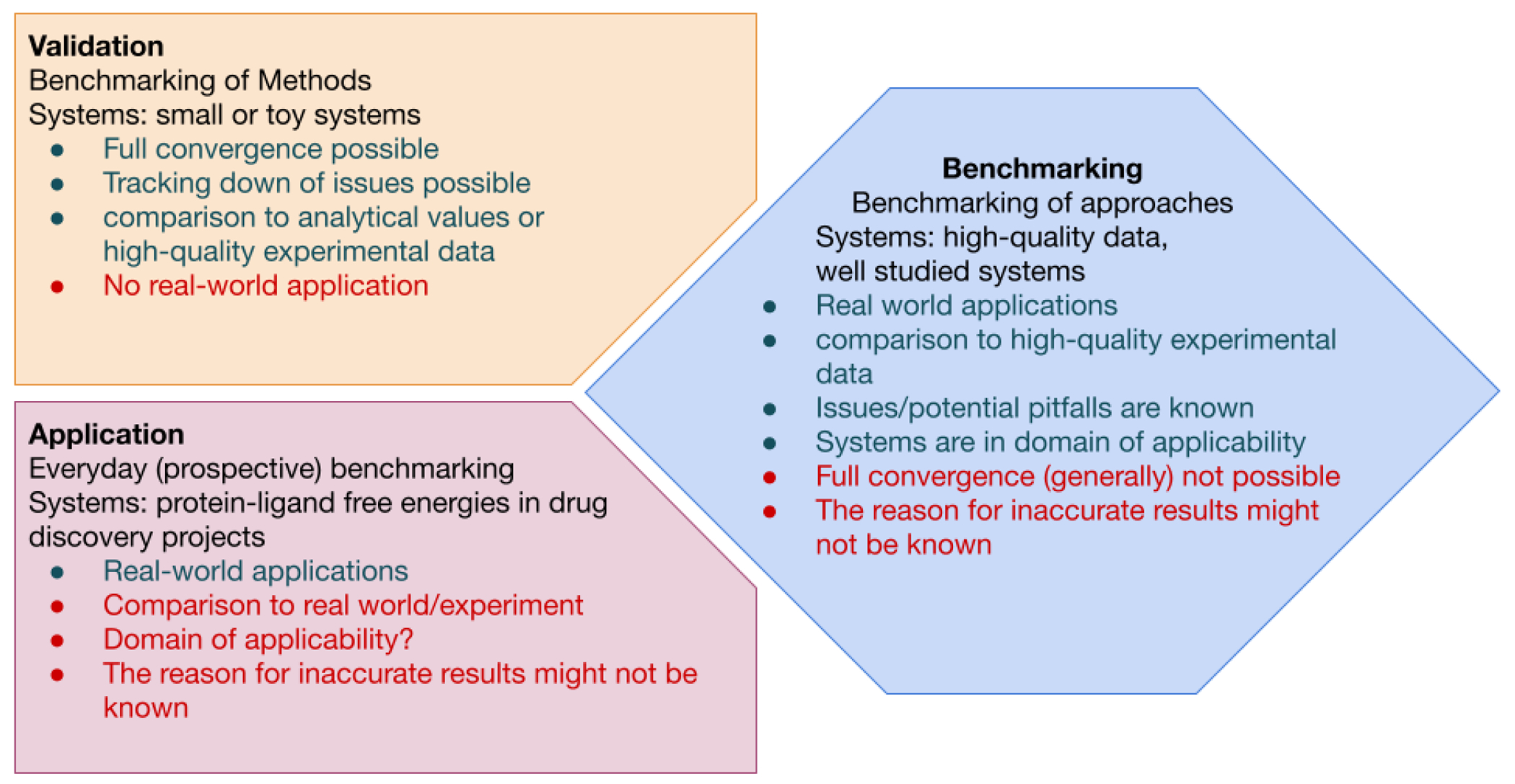
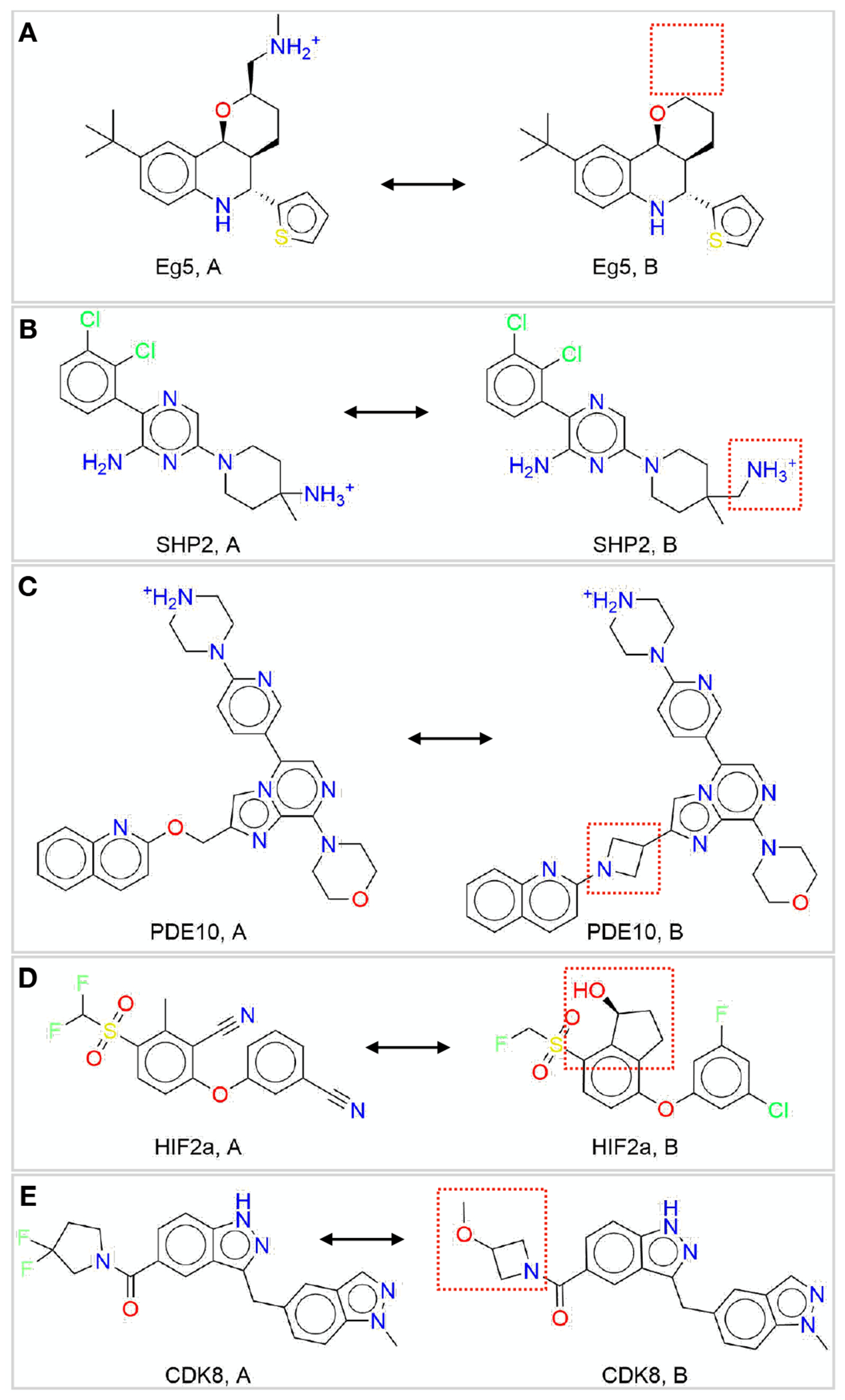
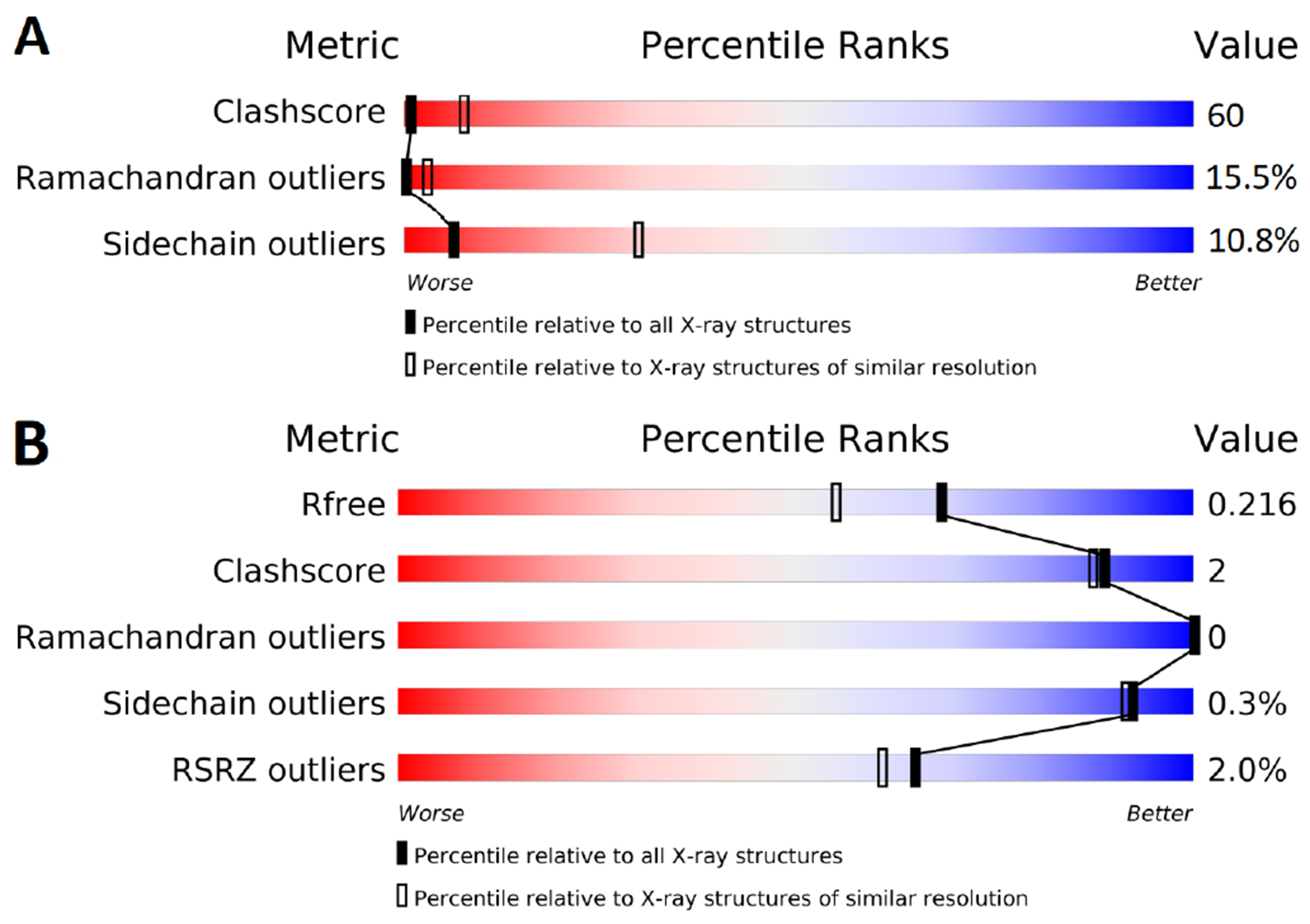
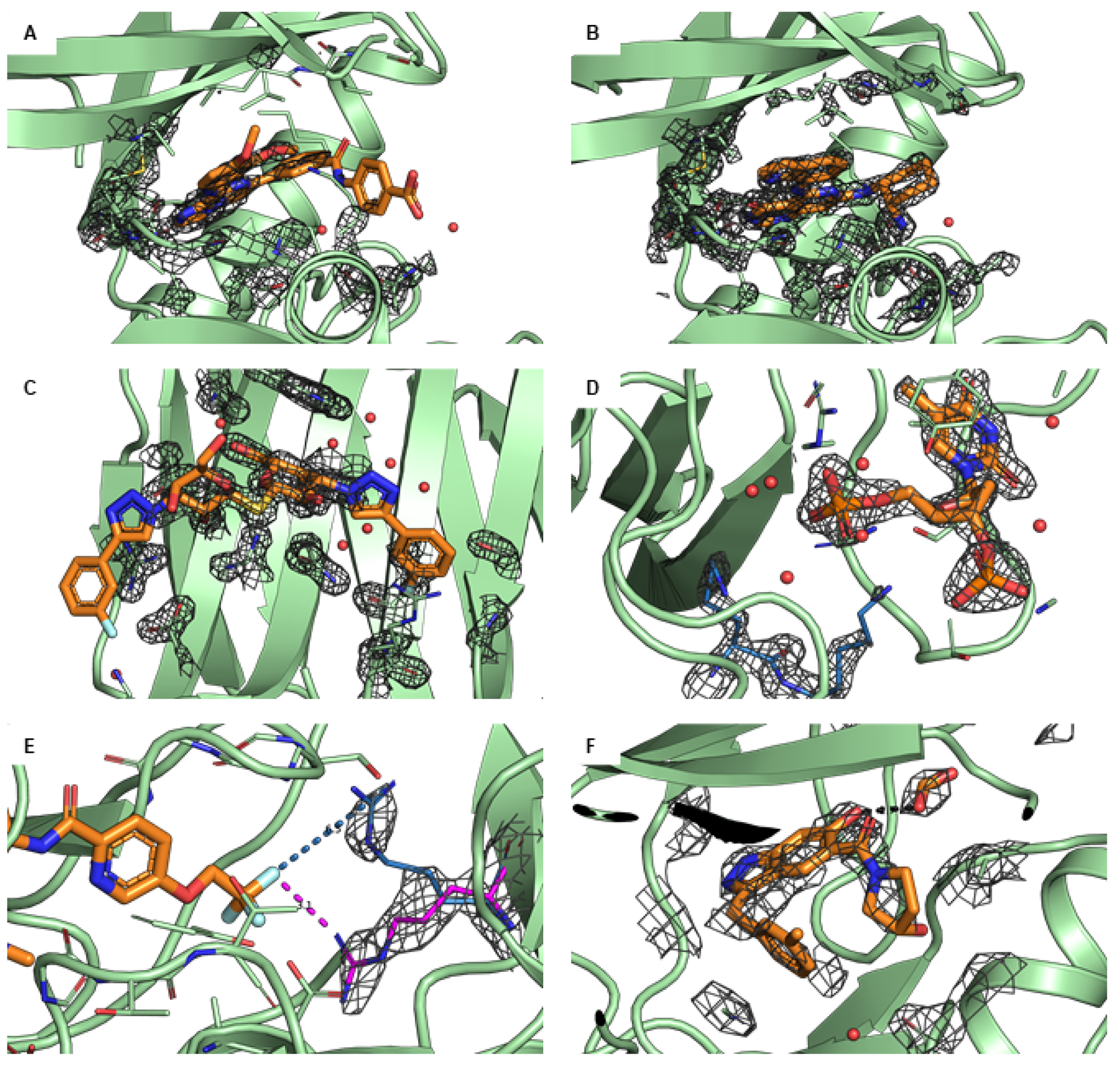
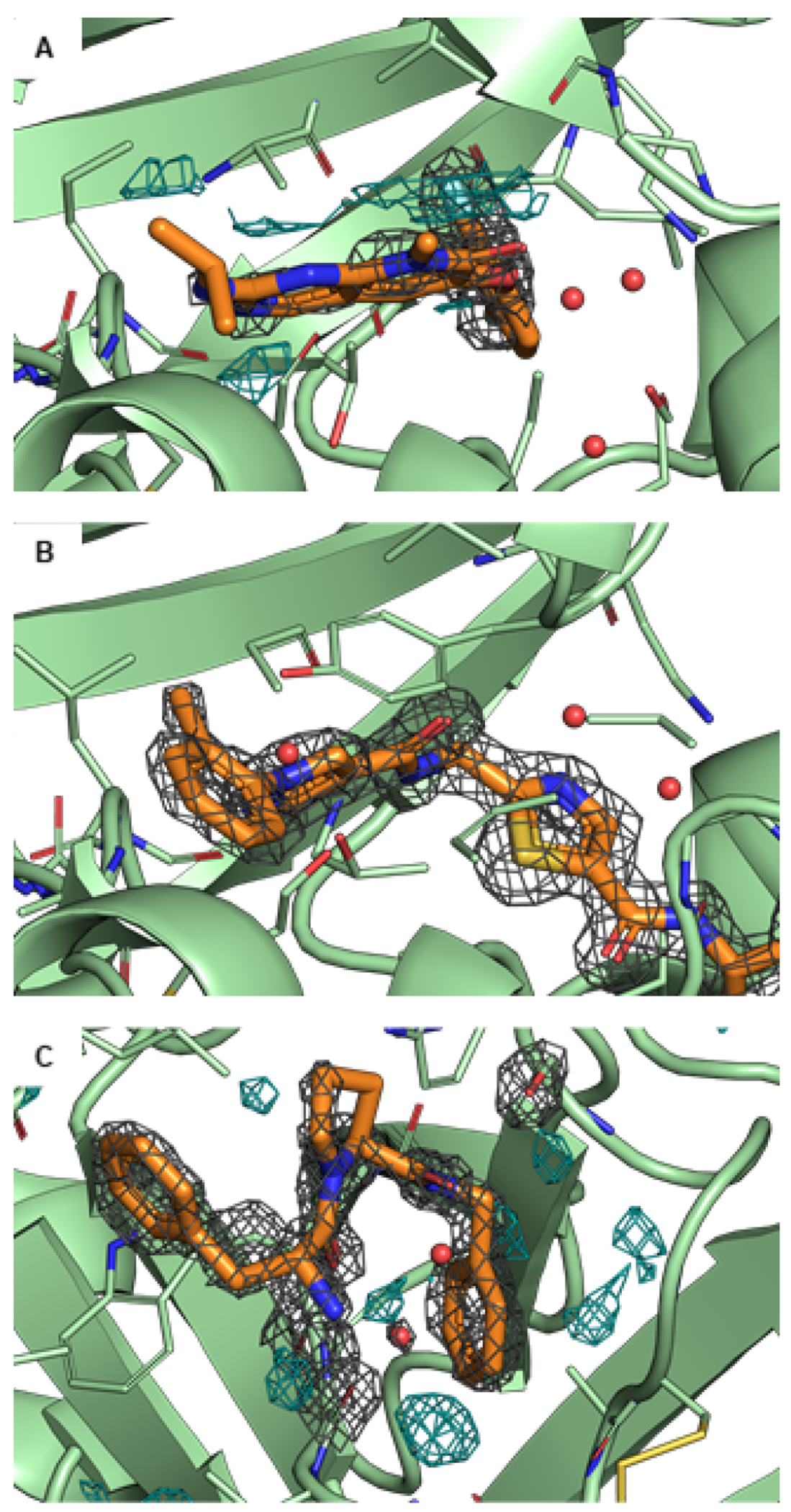
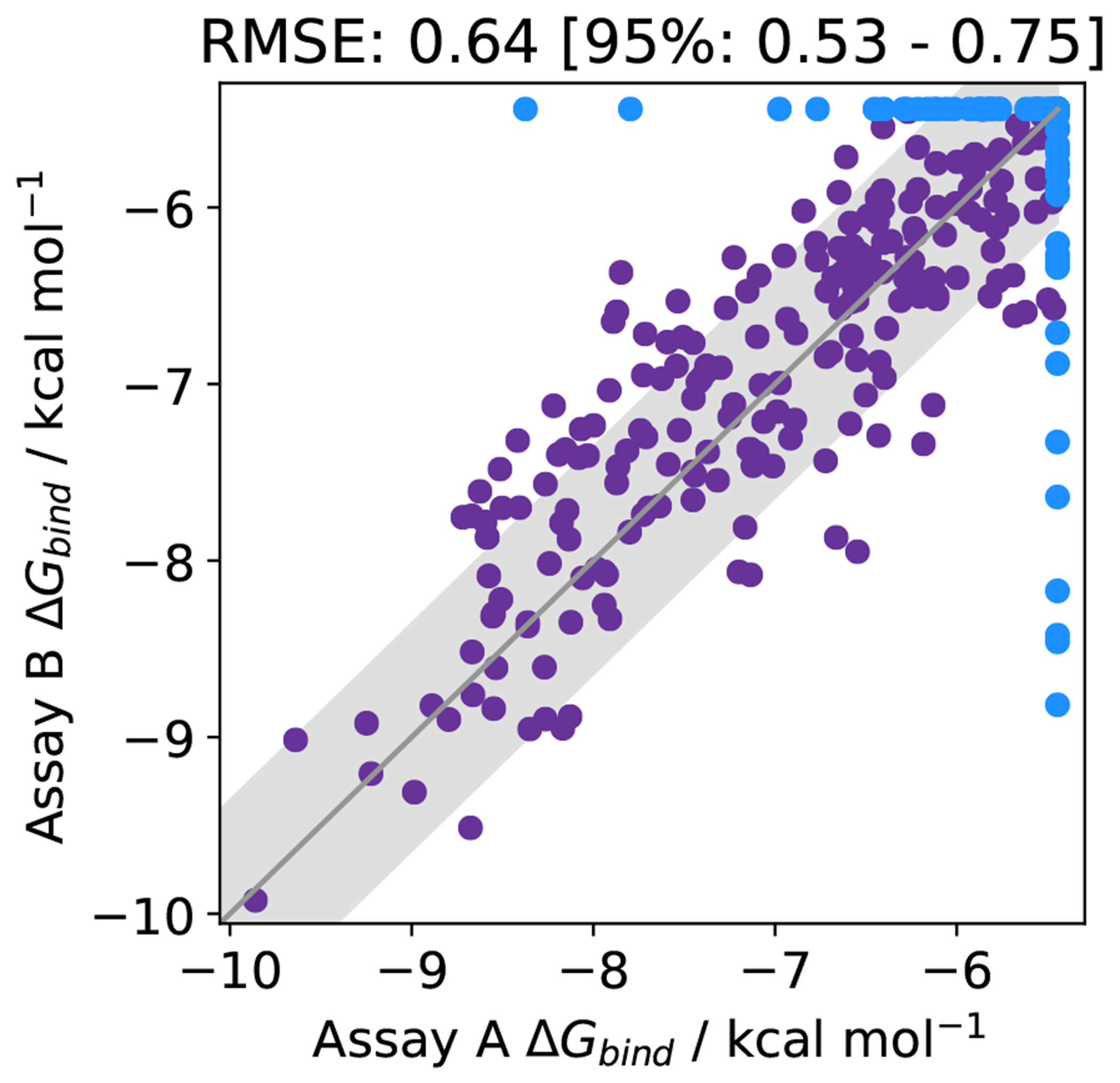
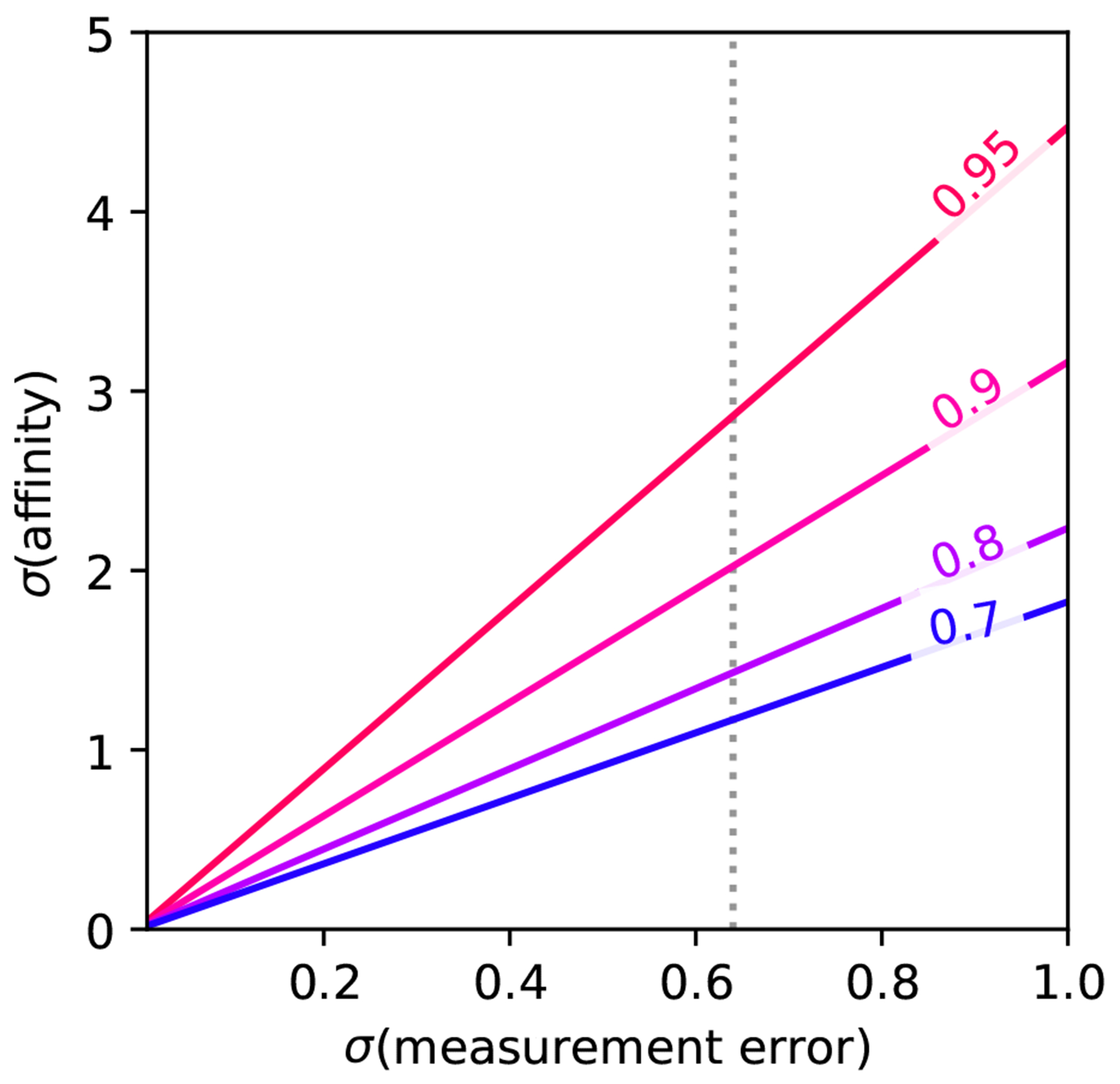
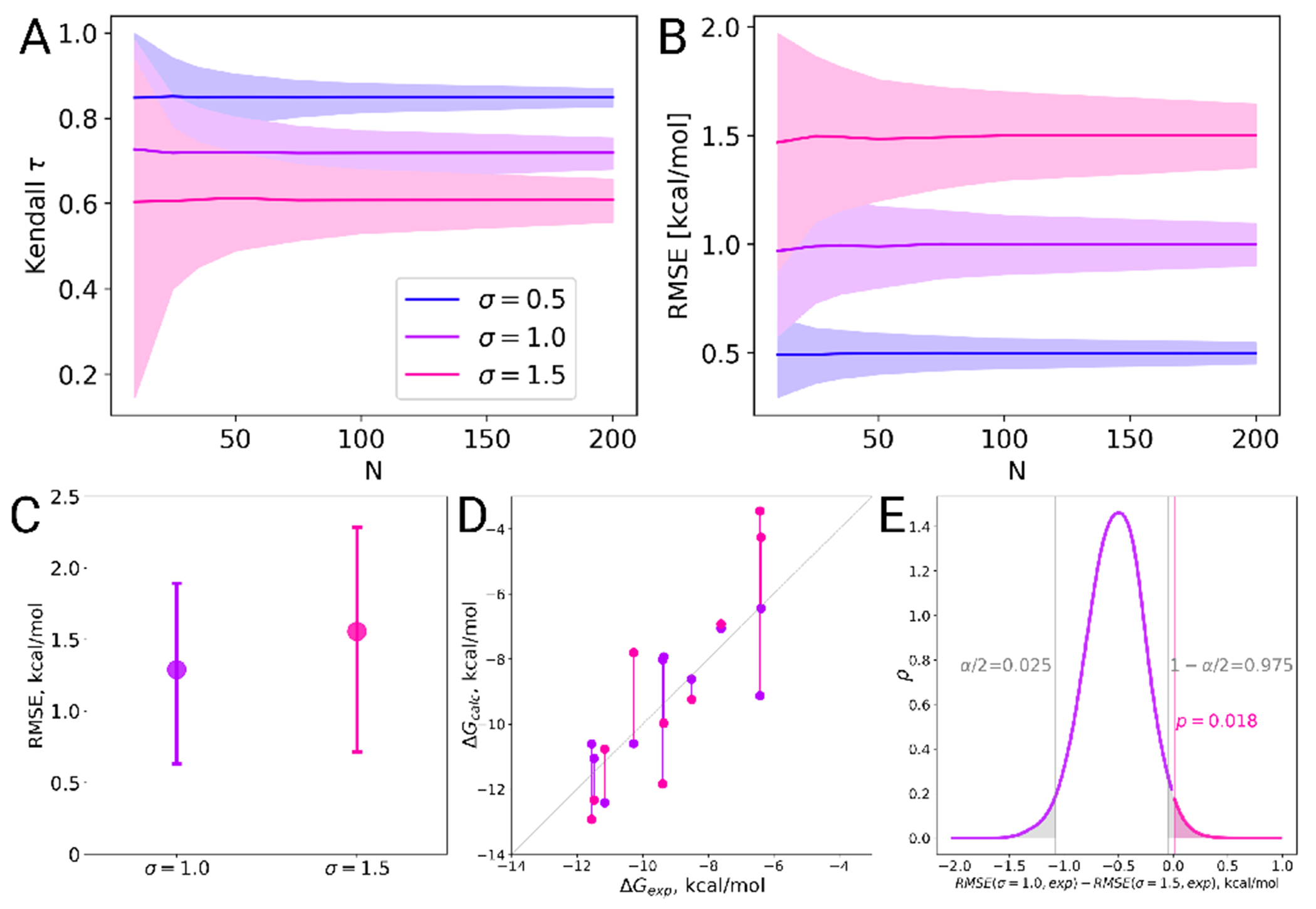
Free energy calculations are rapidly becoming indispensable in structure-enabled drug discovery programs. As new methods, force fields, and implementations are developed, assessing their expected accuracy on real-world systems (benchmarking) becomes critical to provide users with an assessment of the accuracy expected when these methods are applied within their domain of applicability, and developers with a way to assess the expected impact of new methodologies. These assessments require construction of a benchmark-a set of well-prepared, high quality systems with corresponding experimental measurements designed to ensure the resulting calculations provide a realistic assessment of expected performance when these methods are deployed within their domains of applicability. To date, the community has not yet adopted a common standardized benchmark, and existing benchmark reports suffer from a myriad of issues, including poor data quality, limited statistical power, and statistically deficient analyses, all of which can conspire to produce benchmarks that are poorly predictive of real-world performance. Here, we address these issues by presenting guidelines for (1) curating experimental data to develop meaningful benchmark sets, (2) preparing benchmark inputs according to best practices to facilitate widespread adoption, and (3) analysis of the resulting predictions to enable statistically meaningful comparisons among methods and force fields. We highlight challenges and open questions that remain to be solved in these areas, as well as recommendations for the collection of new datasets that might optimally serve to measure progress as methods become systematically more reliable. Finally, we provide a curated, versioned, open, standardized benchmark set adherent to these standards (PLBenchmarks) and an open source toolkit for implementing standardized best practices assessments (arsenic) for the community to use as a standardized assessment tool. While our main focus is free energy methods based on molecular simulations, these guidelines should prove useful for assessment of the rapidly growing field of machine learning methods for affinity prediction as well.
Conflict of interest statement
12Potentially Conflicting Interests DLM serves on the scientific advisory board for OpenEye Scientific Software and is an Open Science Fellow with Silicon Therapeutics. ASJSM is a consultant for Exscientia. JDC is a current member of the Scientific Advisory Board of OpenEye Scientific Software and a consultant to Foresite Laboratories. HEBM is employed by MSD.
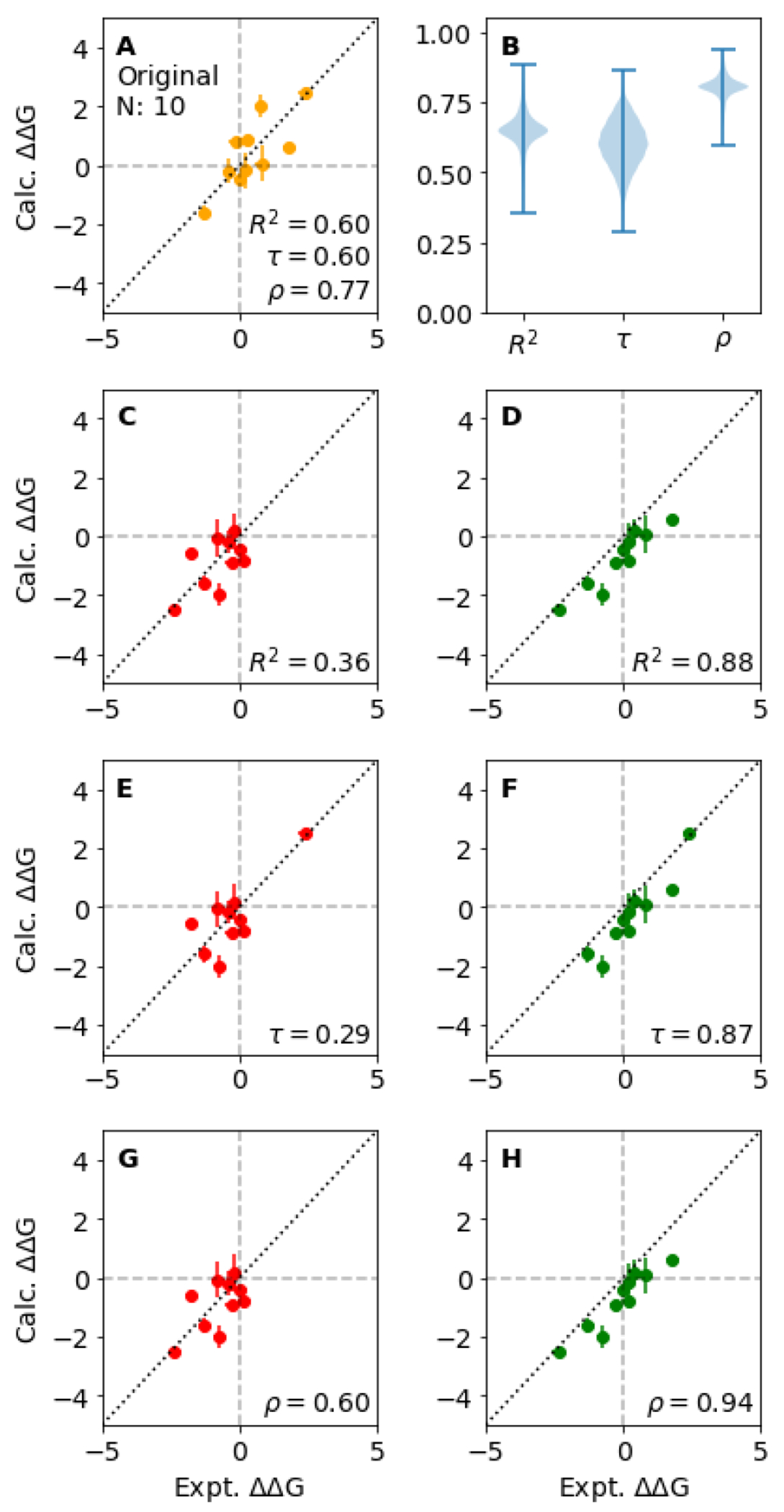
Figures
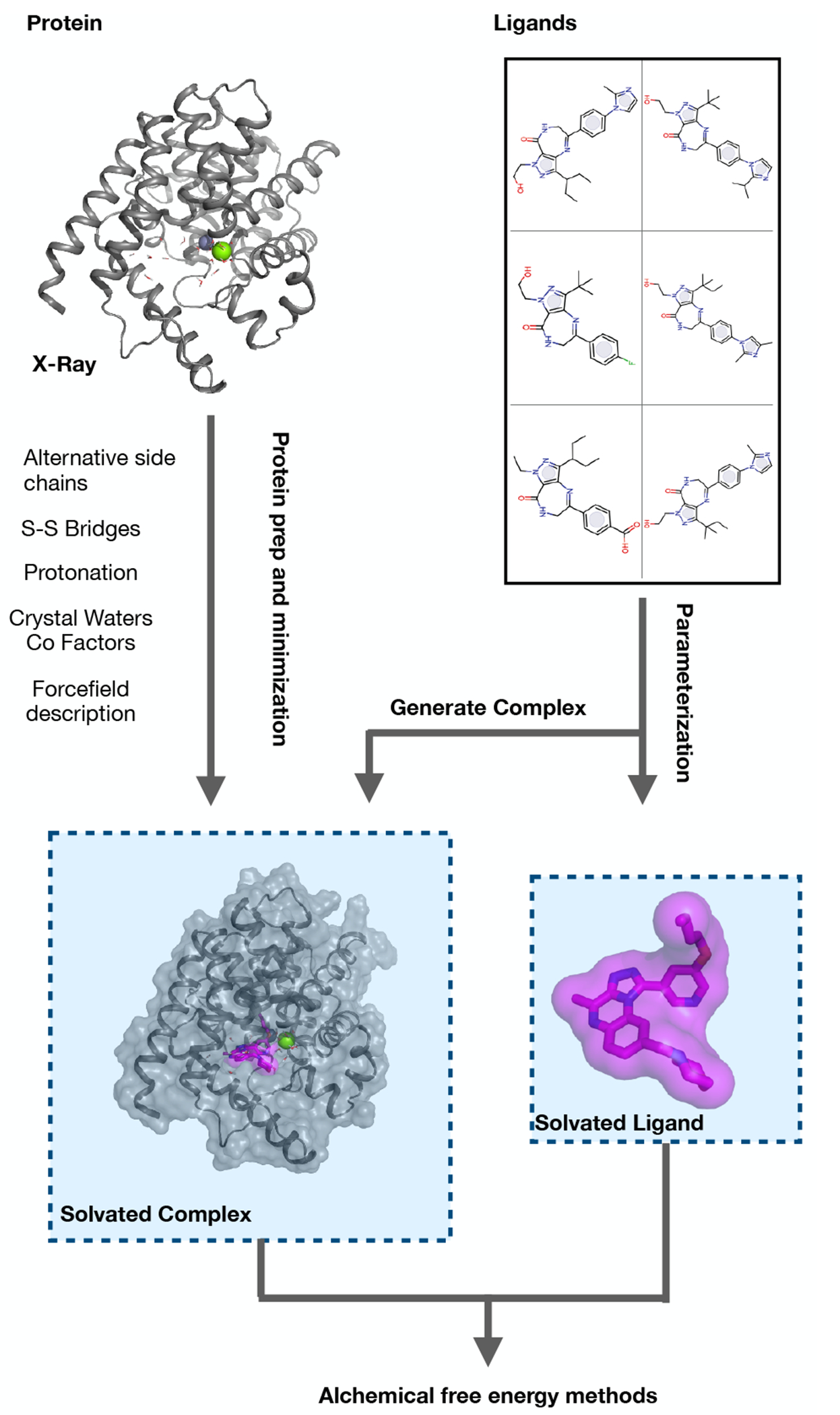
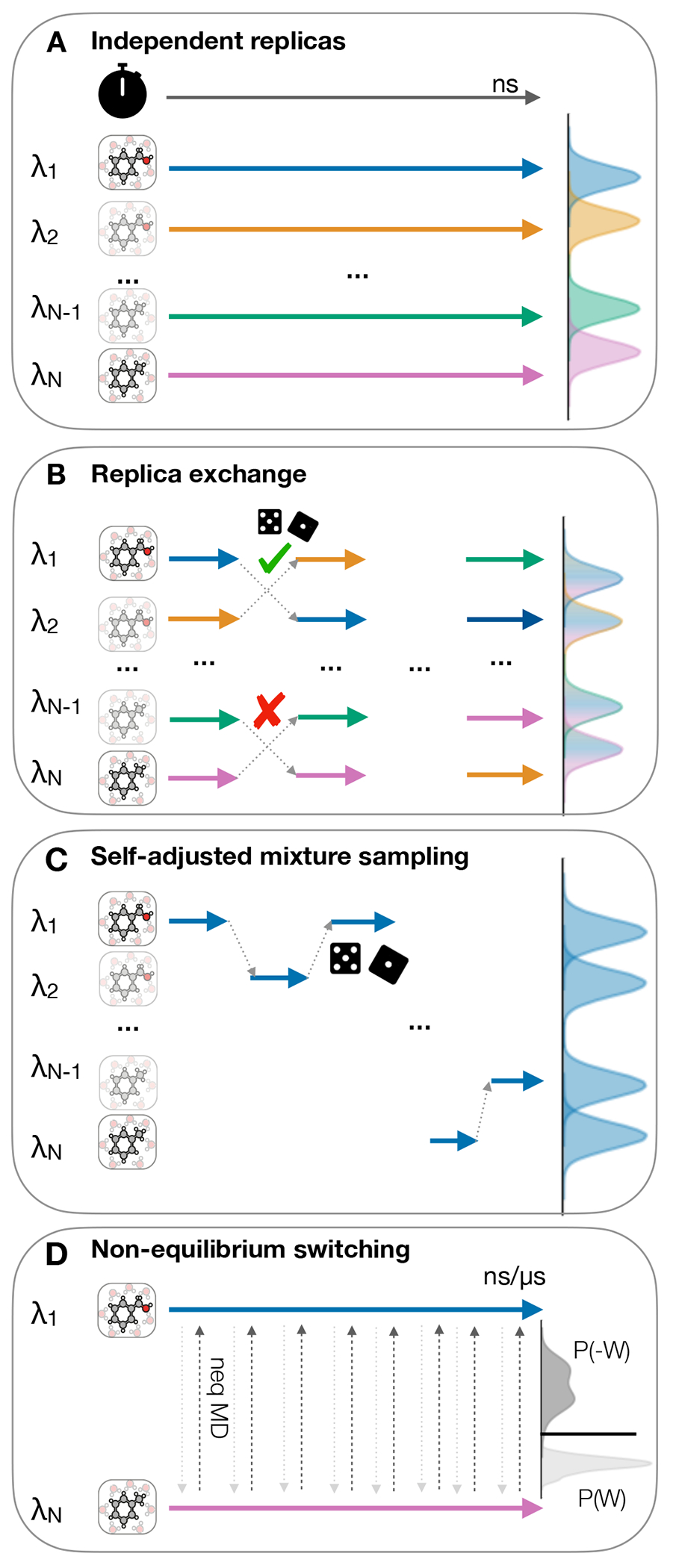
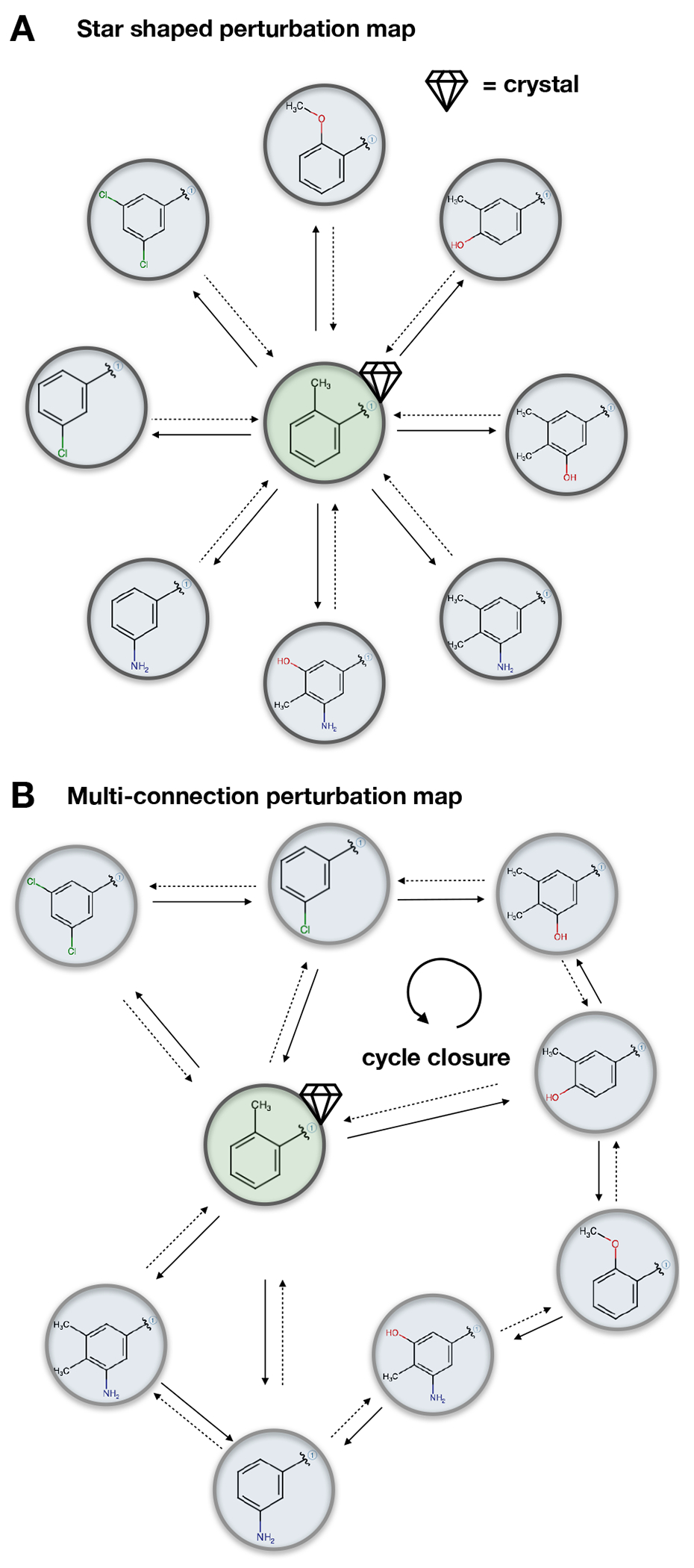
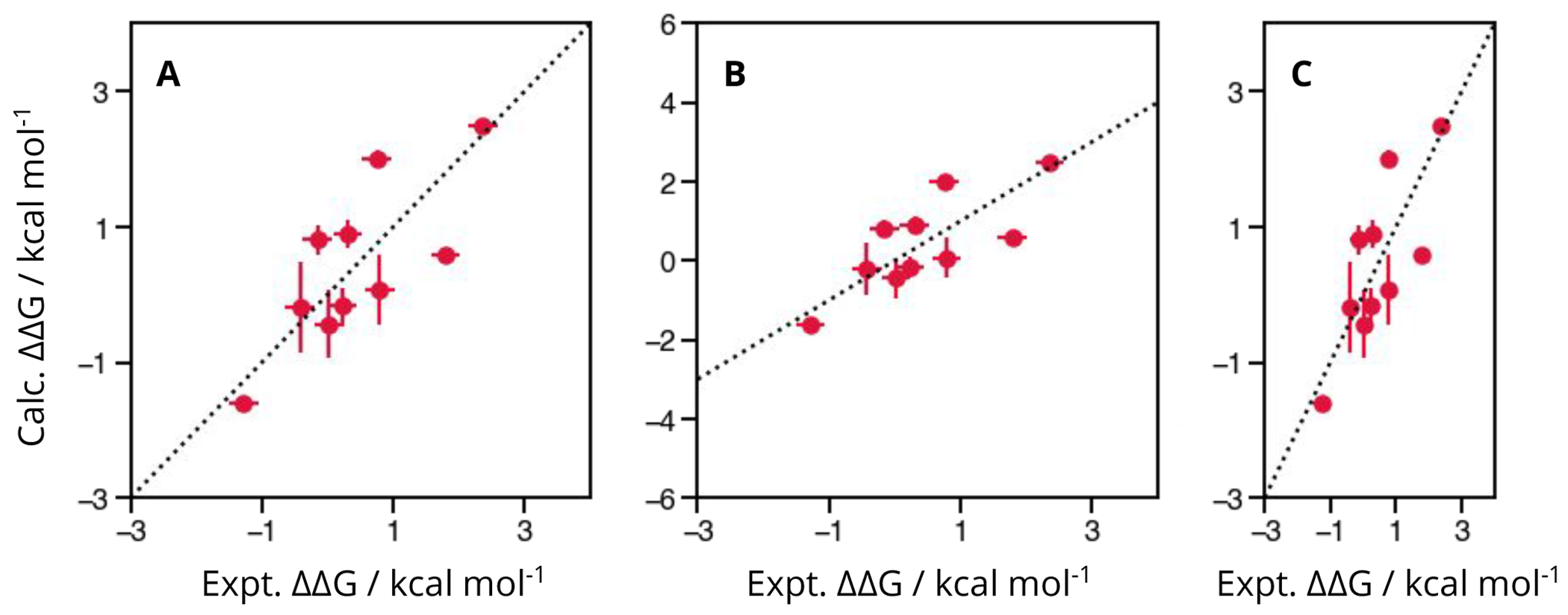
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials