

Overview and considerations in bottom-up proteomics
- PMID: 36383138
- PMCID: PMC9898146
- DOI: 10.1039/d2an01246d
Overview and considerations in bottom-up proteomics
Abstract
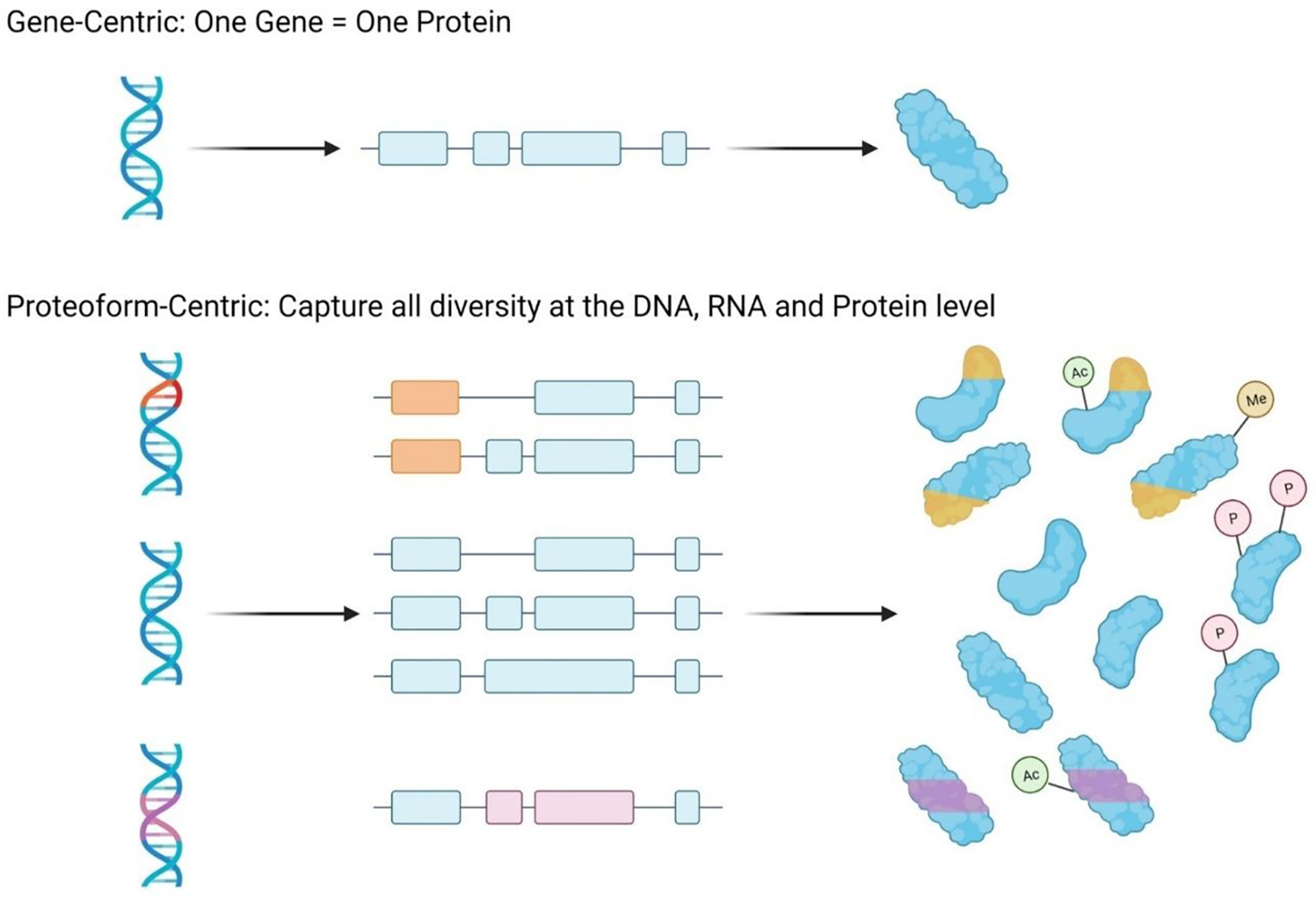
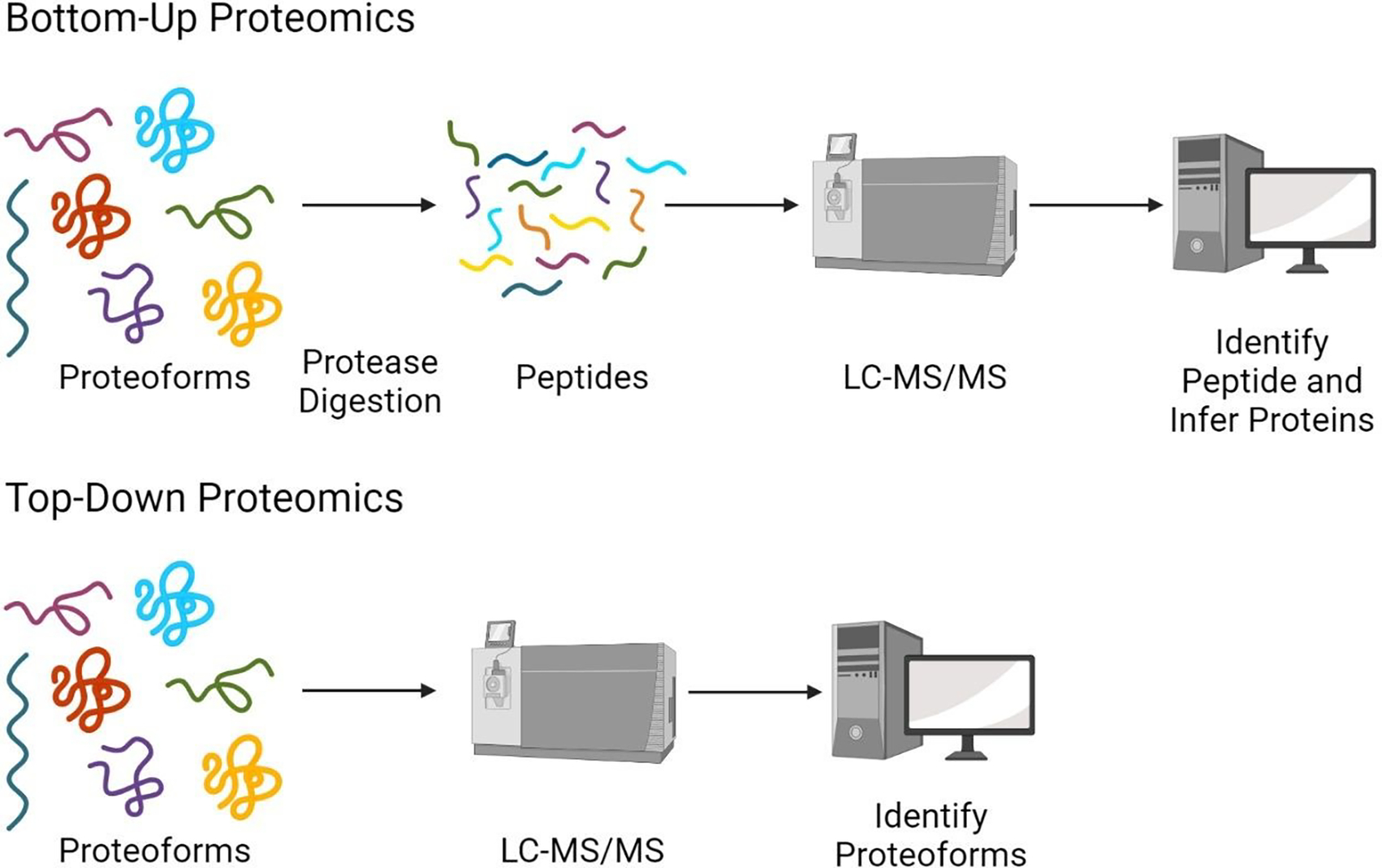
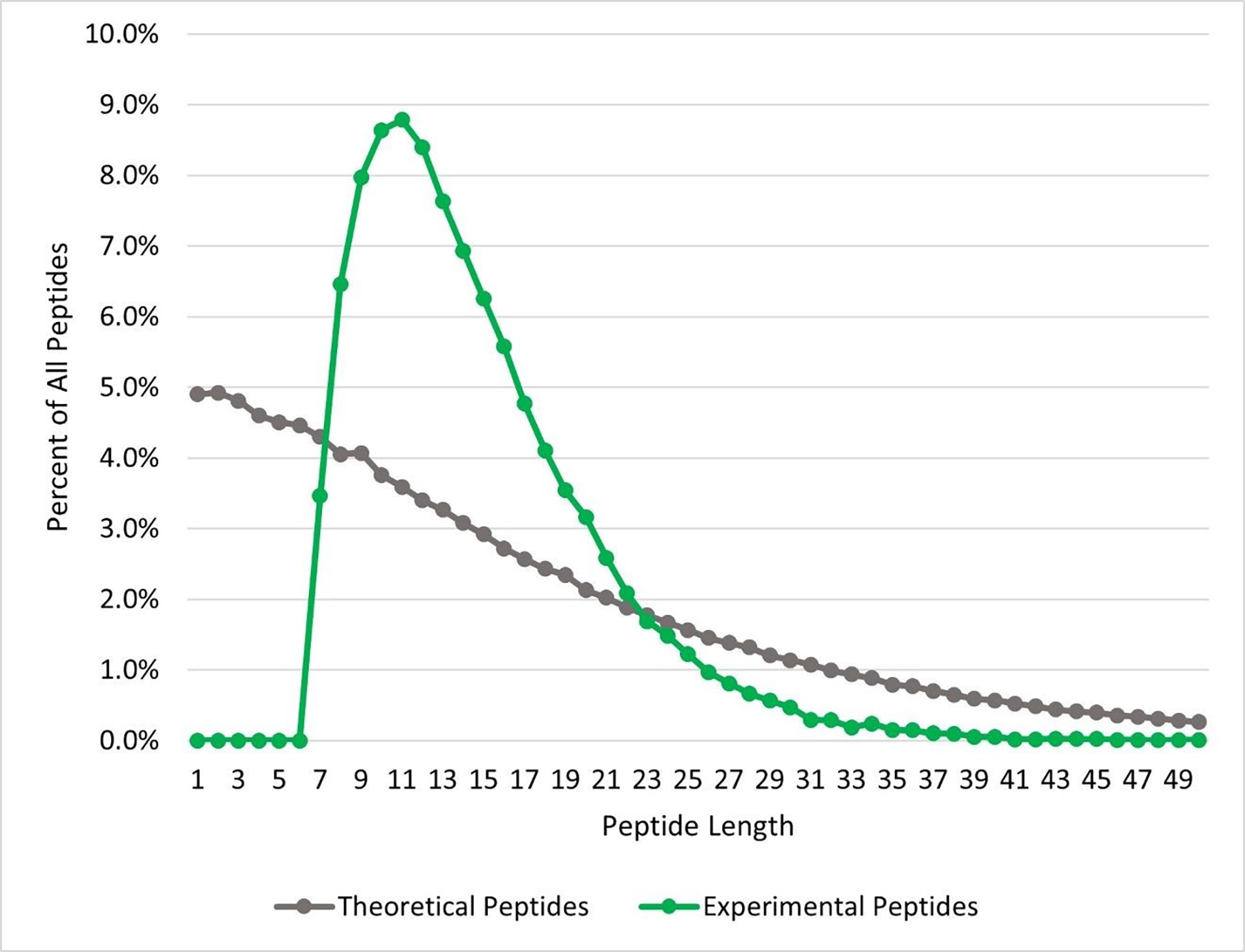
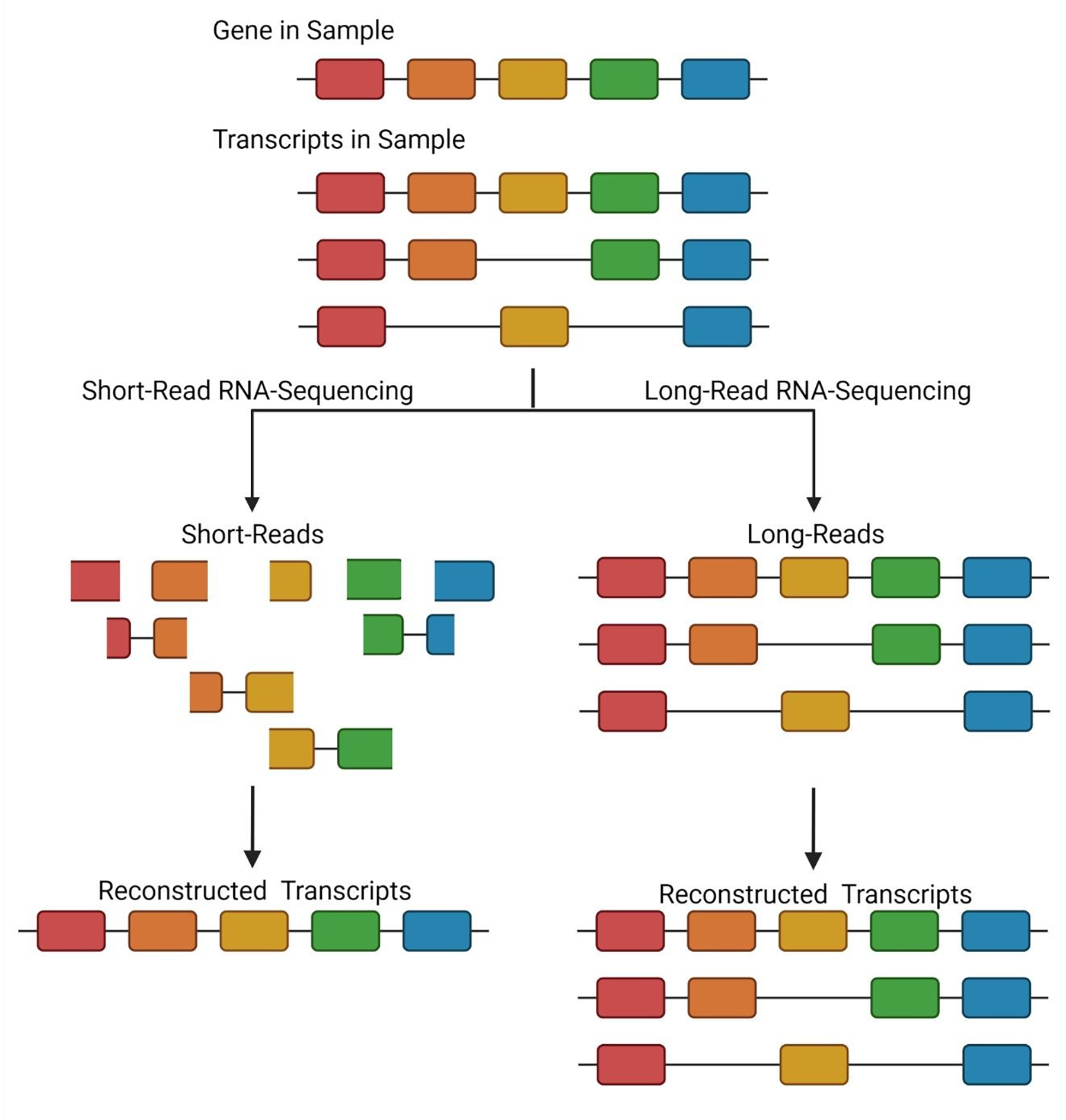
Proteins are the key biological actors within cells, driving many biological processes integral to both healthy and diseased states. Understanding the depth of complexity represented within the proteome is crucial to our scientific understanding of cellular biology and to provide disease specific insights for clinical applications. Mass spectrometry-based proteomics is the premier method for proteome analysis, with the ability to both identify and quantify proteins. Although proteomics continues to grow as a robust field of bioanalytical chemistry, advances are still necessary to enable a more comprehensive view of the proteome. In this review, we provide a broad overview of mass spectrometry-based proteomics in general, and highlight four developing areas of bottom-up proteomics: (1) protein inference, (2) alternative proteases, (3) sample-specific databases and (4) post-translational modification discovery.
Figures
References
-
- Aebersold R; Agar JN; Amster IJ; Baker MS; Bertozzi CR; Boja ES; Costello CE; Cravatt BF; Fenselau C; Garcia BA; Ge Y; Gunawardena J; Hendrickson RC; Hergenrother PJ; Huber CG; Ivanov AR; Jensen ON; Jewett MC; Kelleher NL; Kiessling LL; Krogan NJ; Larsen MR; Loo JA; Ogorzalek Loo RR; Lundberg E; MacCoss MJ; Mallick P; Mootha VK; Mrksich M; Muir TW; Patrie SM; Pesavento JJ; Pitteri SJ; Rodriguez H; Saghatelian A; Sandoval W; Schlüter H; Sechi S; Slavoff SA; Smith LM; Snyder MP; Thomas PM; Uhlén M; Van Eyk JE; Vidal M; Walt DR; White FM; Williams ER; Wohlschlager T; Wysocki VH; Yates NA; Young NL; Zhang B How Many Human Proteoforms Are There? Nat. Chem. Biol. 2018, 14 (3), 206–214. 10.1038/nchembio.2576. - DOI - PMC - PubMed
-
- Uhlén M; Fagerberg L; Hallström BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson Å; Kampf C; Sjöstedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA-K; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist P-H; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Pontén F Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347 (6220), 1260419. 10.1126/science.1260419. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources