

Annotation of structural variants with reported allele frequencies and related metrics from multiple datasets using SVAFotate
- PMID: 36384437
- PMCID: PMC9670370
- DOI: 10.1186/s12859-022-05008-y
Annotation of structural variants with reported allele frequencies and related metrics from multiple datasets using SVAFotate
Abstract
Background: Identification of deleterious genetic variants using DNA sequencing data relies on increasingly detailed filtering strategies to isolate the small subset of variants that are more likely to underlie a disease phenotype. Datasets reflecting population allele frequencies of different types of variants serve as powerful filtering tools, especially in the context of rare disease analysis. While such population-scale allele frequency datasets now exist for structural variants (SVs), it remains a challenge to match SV calls between multiple datasets, thereby complicating estimates of a putative SV's population allele frequency.
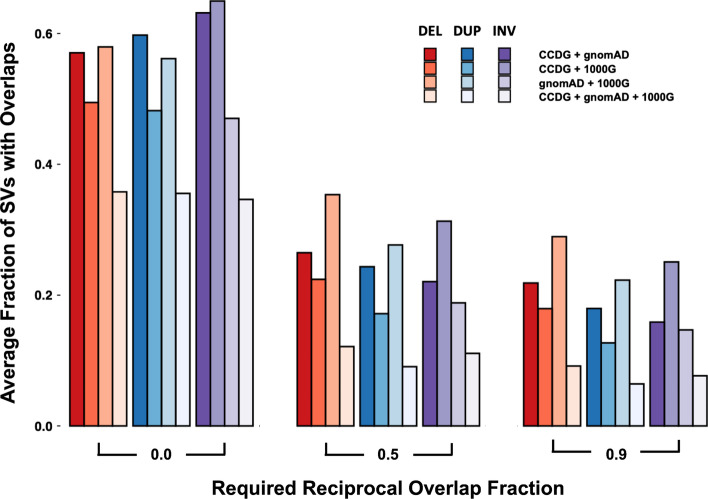
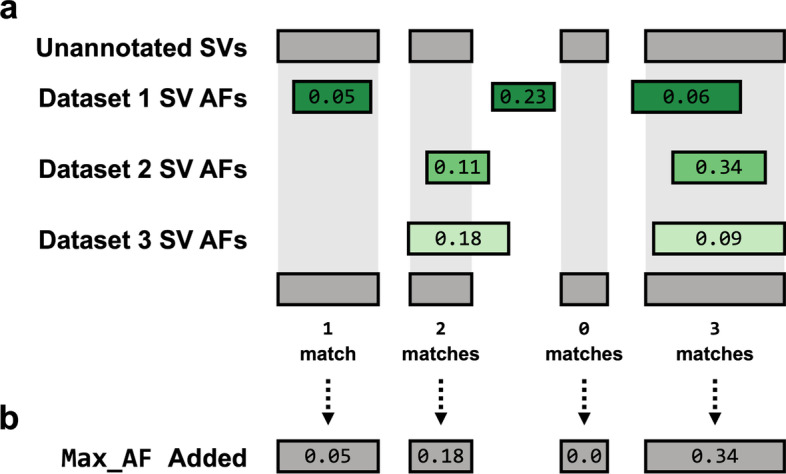
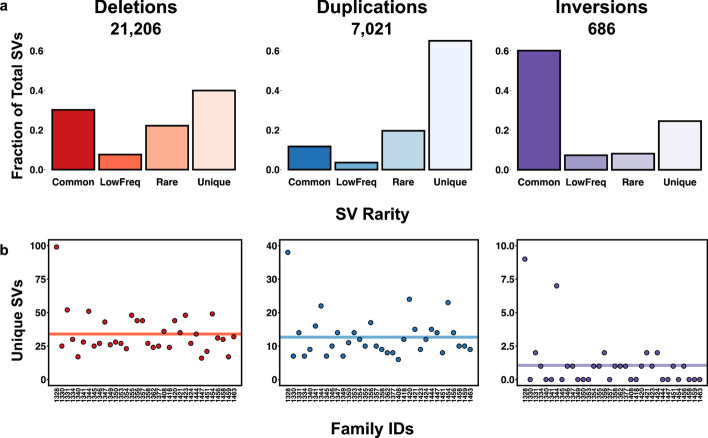
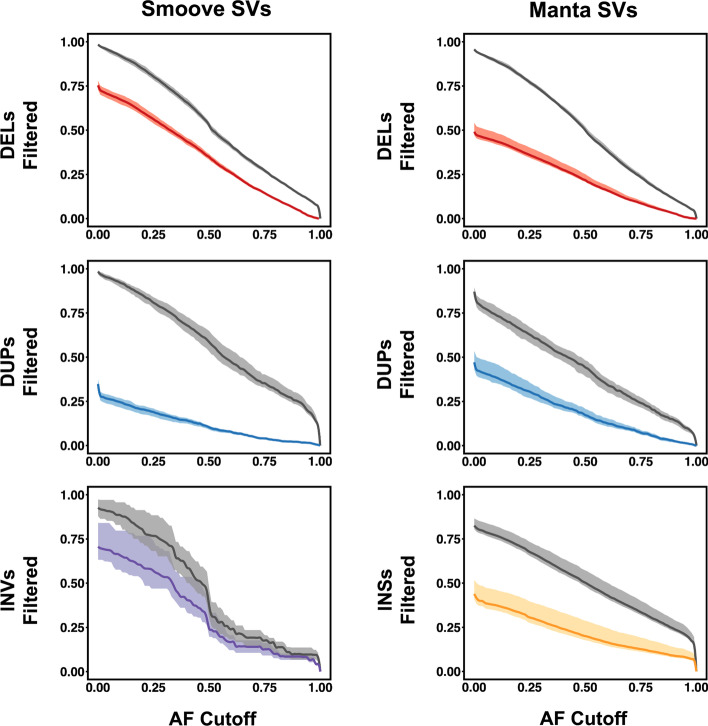
Results: We introduce SVAFotate, a software tool that enables the annotation of SVs with variant allele frequency and related information from existing SV datasets. As a result, VCF files annotated by SVAFotate offer a variety of metrics to aid in the stratification of SVs as common or rare in the broader human population.
Conclusions: Here we demonstrate the use of SVAFotate in the classification of SVs with regards to their population frequency and illustrate how SVAFotate's annotations can be used to filter and prioritize SVs. Lastly, we detail how best to utilize these SV annotations in the analysis of genetic variation in studies of rare disease.
Keywords: Genome annotation; Population allele frequency; Structural variation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
