

lmerSeq: an R package for analyzing transformed RNA-Seq data with linear mixed effects models
- PMID: 36384492
- PMCID: PMC9670578
- DOI: 10.1186/s12859-022-05019-9
lmerSeq: an R package for analyzing transformed RNA-Seq data with linear mixed effects models
Abstract
Background: Studies that utilize RNA Sequencing (RNA-Seq) in conjunction with designs that introduce dependence between observations (e.g. longitudinal sampling) require specialized analysis tools to accommodate this additional complexity. This R package contains a set of utilities to fit linear mixed effects models to transformed RNA-Seq counts that properly account for this dependence when performing statistical analyses.
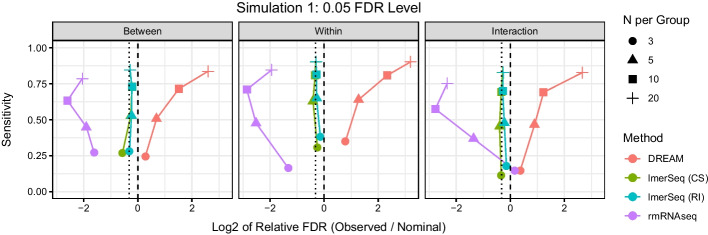
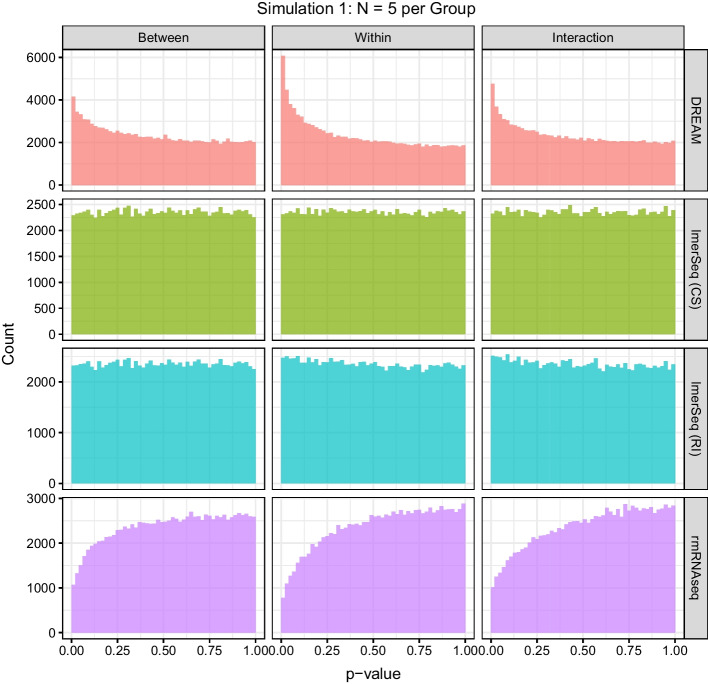
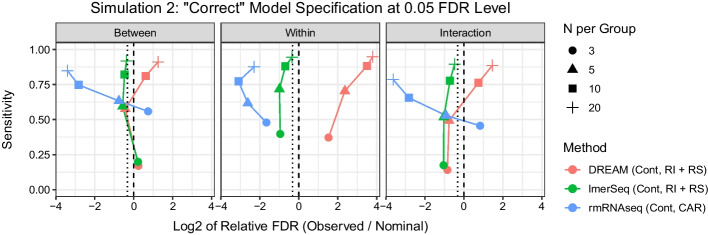
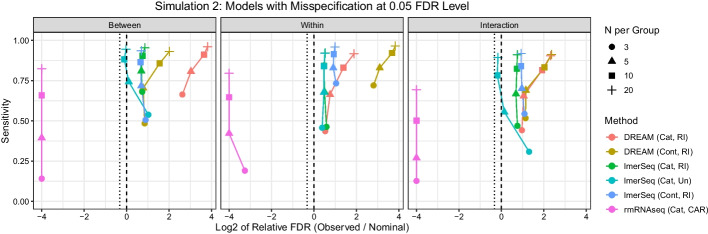
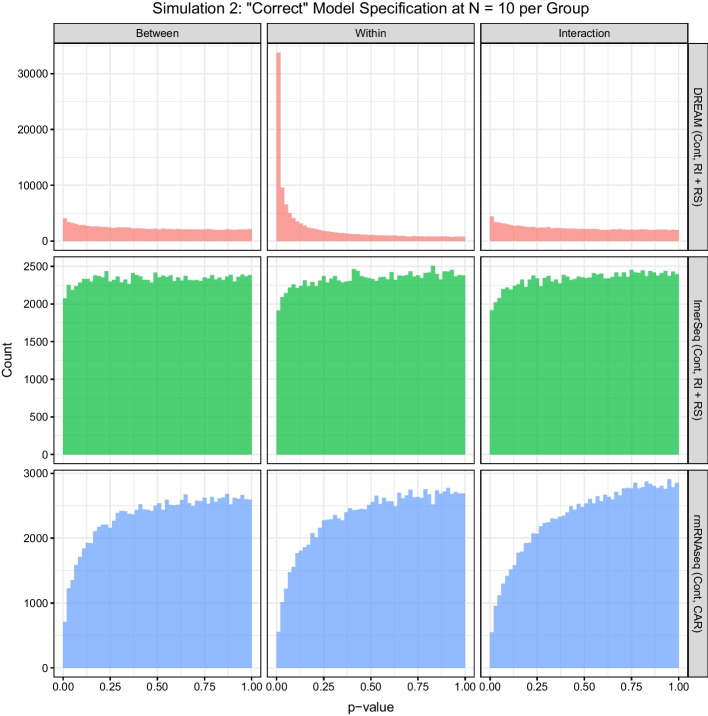
Results: In a simulation study comparing lmerSeq and two existing methodologies that also work with transformed RNA-Seq counts, we found that lmerSeq was comprehensively better in terms of nominal error rate control and statistical power.
Conclusions: Existing R packages for analyzing transformed RNA-Seq data with linear mixed models are limited in the variance structures they allow and/or the transformation methods they support. The lmerSeq package offers more flexibility in both of these areas and gave substantially better results in our simulations.
Keywords: Correlated data; Linear mixed models; RNA-Seq.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
