

PepSeq: a fully in vitro platform for highly multiplexed serology using customizable DNA-barcoded peptide libraries
- PMID: 36385198
- PMCID: PMC10339795
- DOI: 10.1038/s41596-022-00766-8
PepSeq: a fully in vitro platform for highly multiplexed serology using customizable DNA-barcoded peptide libraries
Erratum in
-
Author Correction: PepSeq: a fully in vitro platform for highly multiplexed serology using customizable DNA-barcoded peptide libraries.Nat Protoc. 2024 Nov;19(11):3456. doi: 10.1038/s41596-024-01010-1. Nat Protoc. 2024. PMID: 38755448 No abstract available.
Abstract
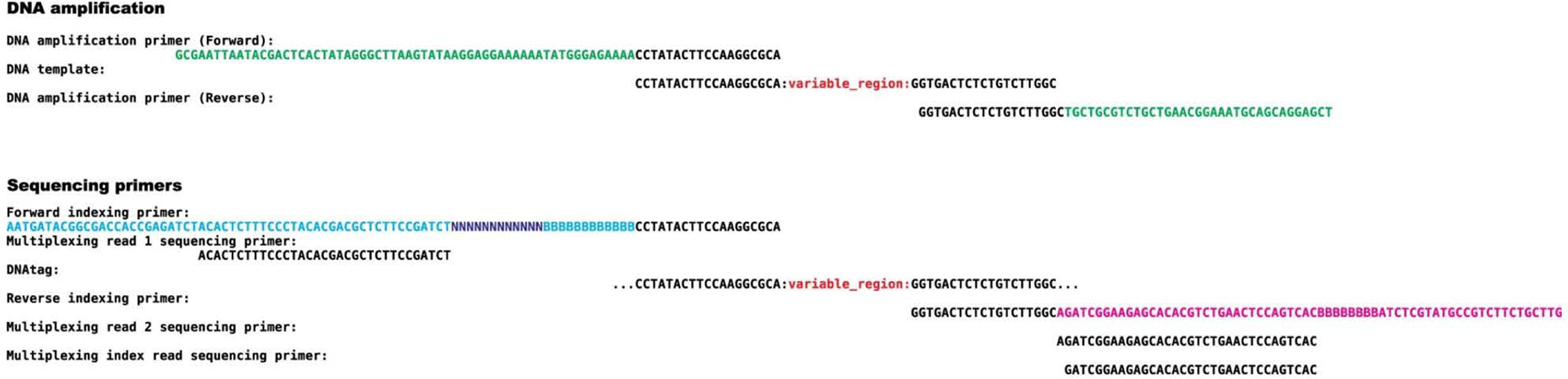
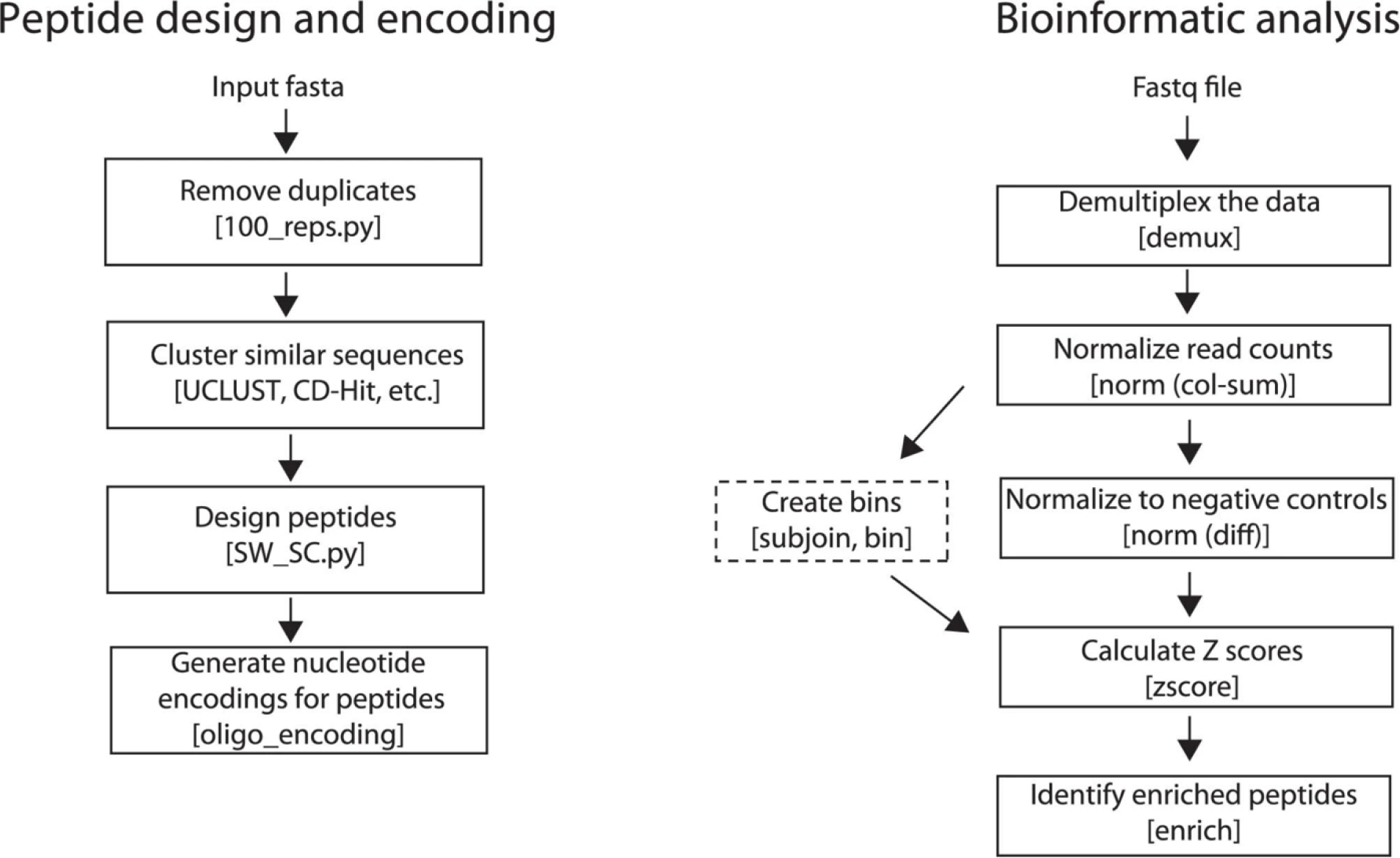
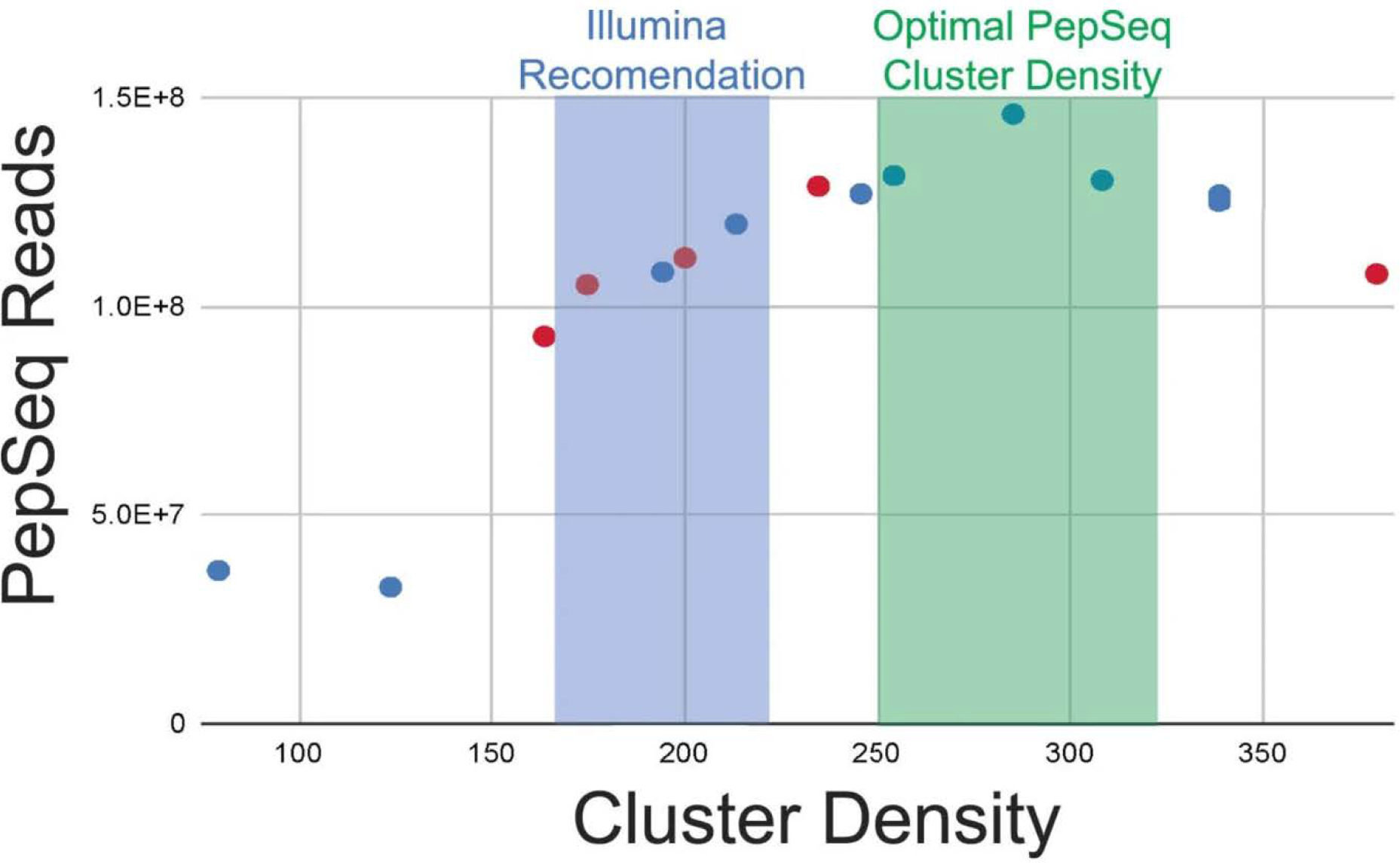
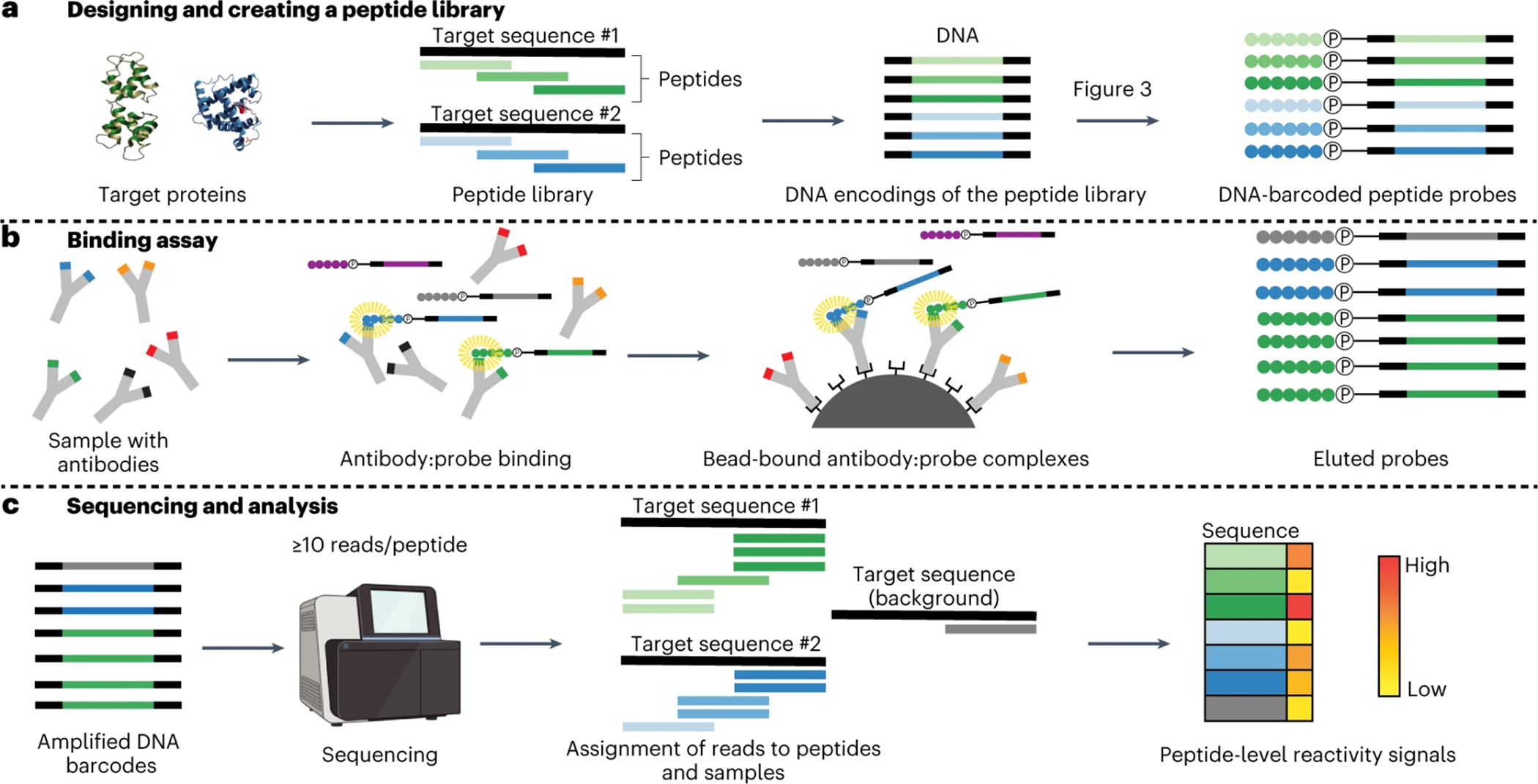
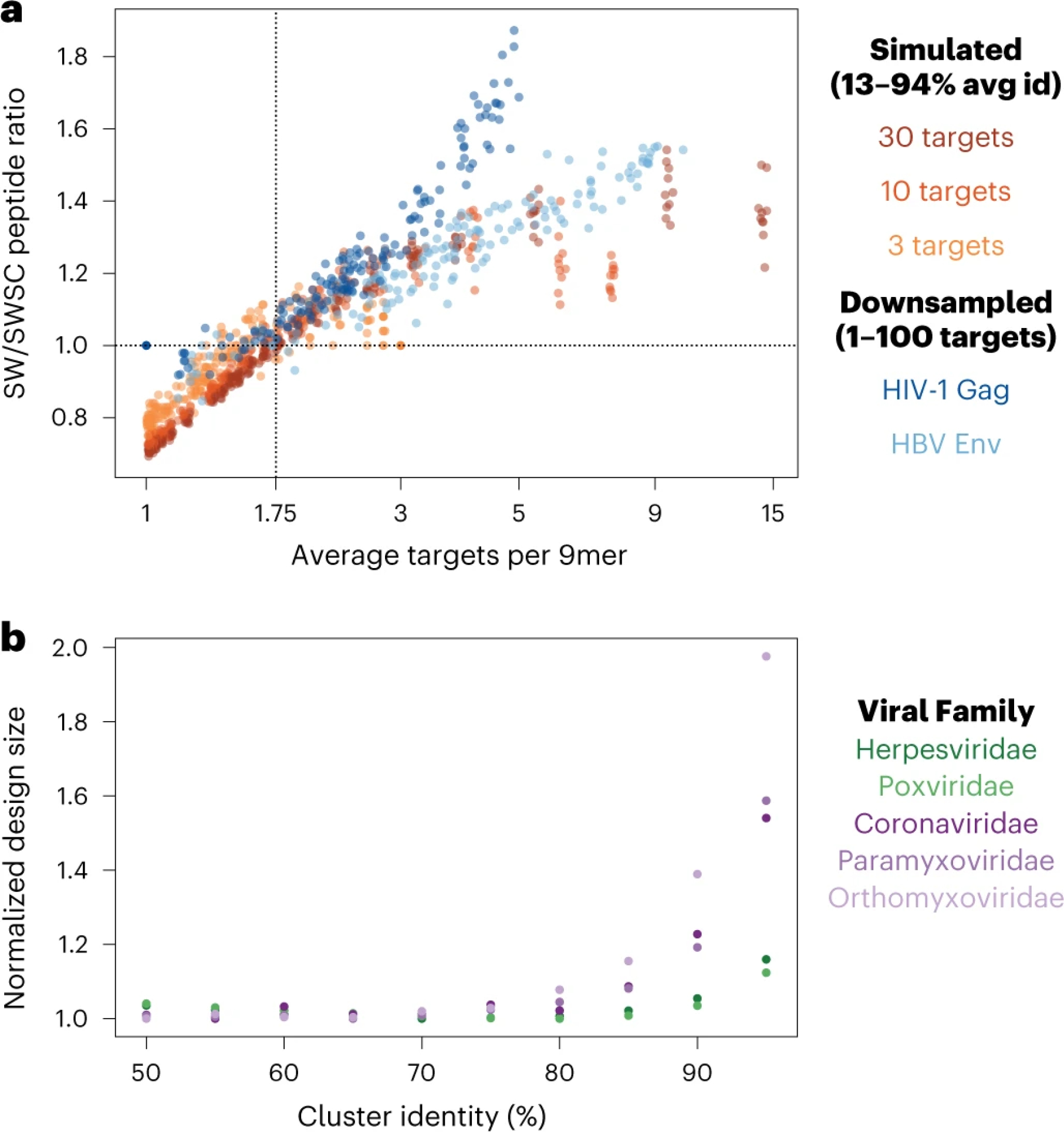
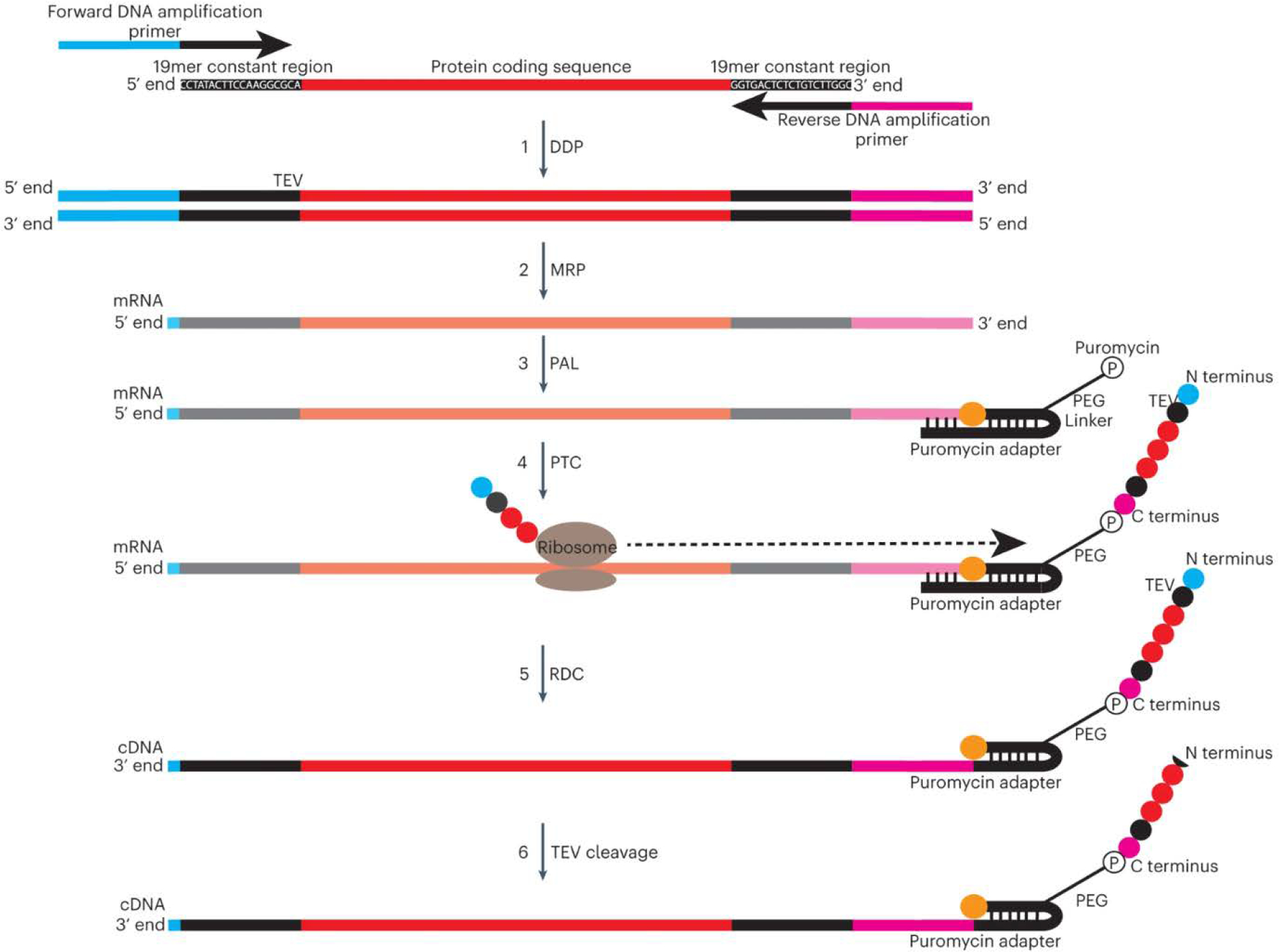
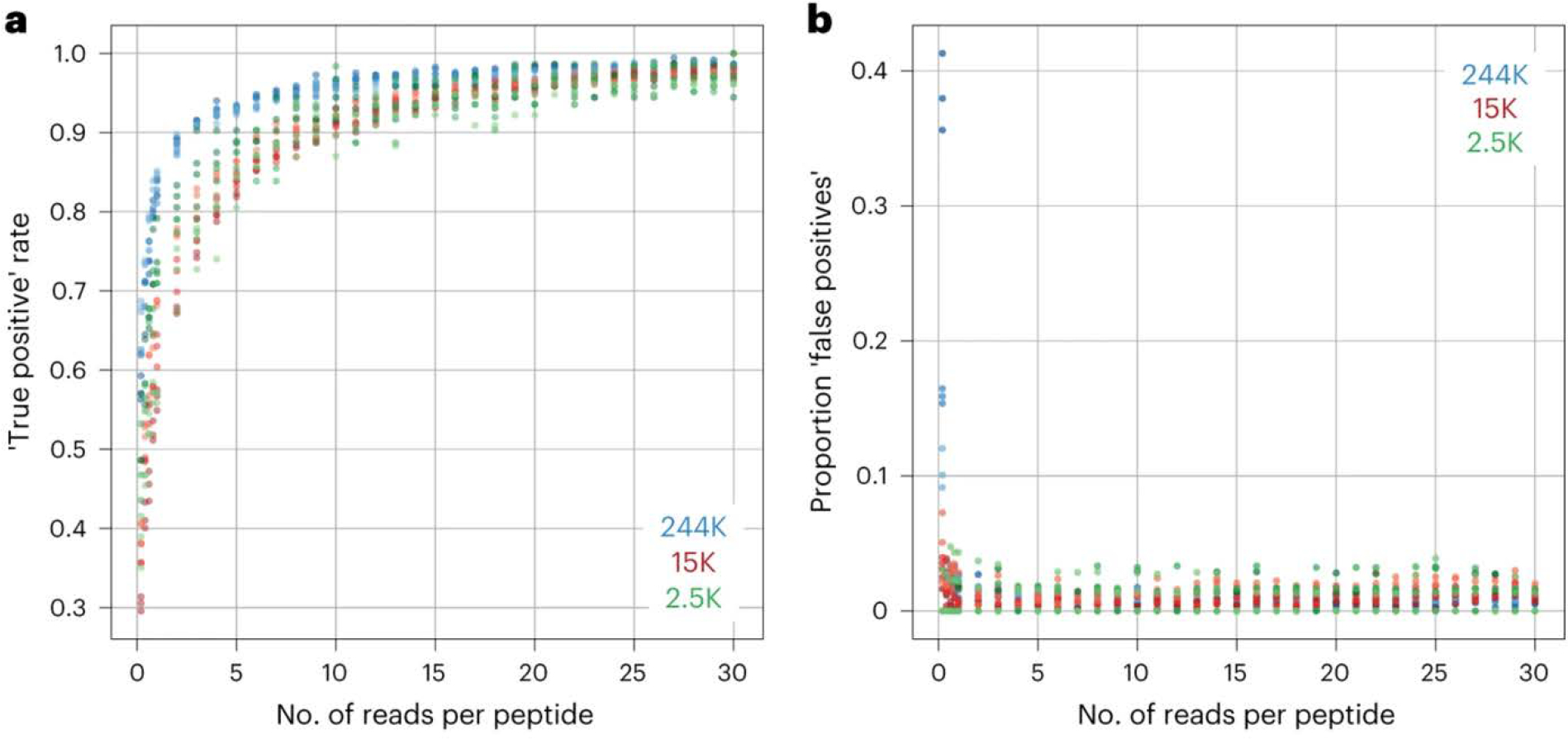
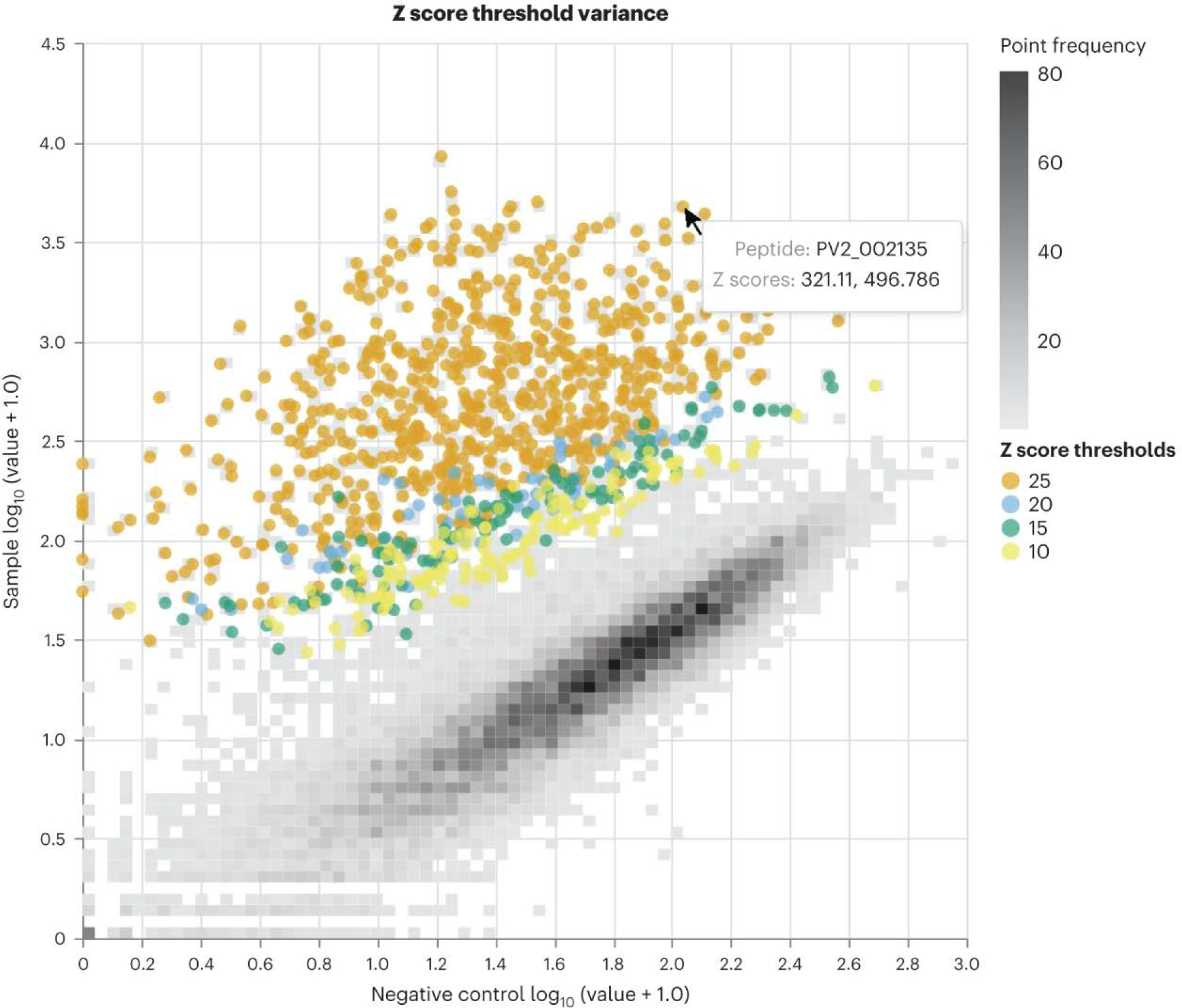
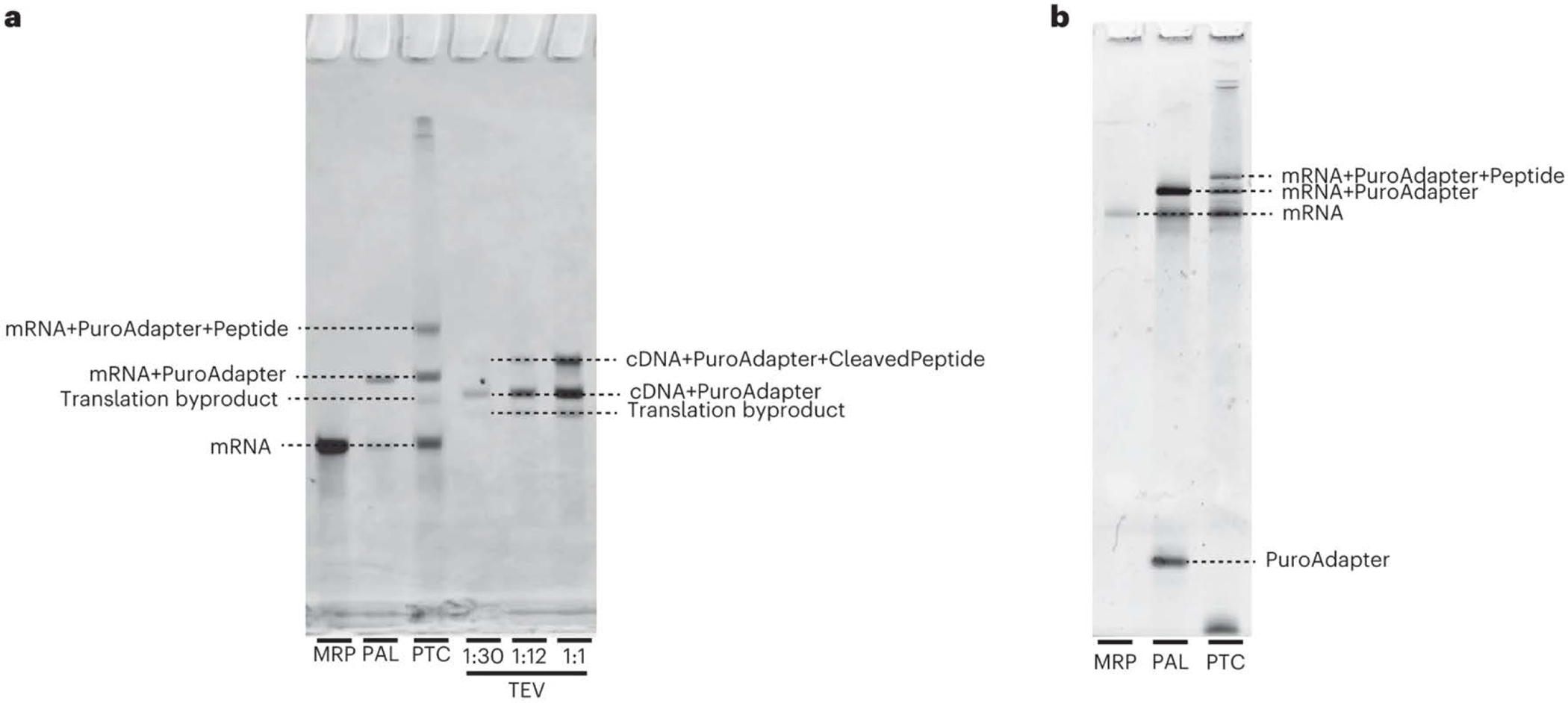
PepSeq is an in vitro platform for building and conducting highly multiplexed proteomic assays against customizable targets by using DNA-barcoded peptides. Starting with a pool of DNA oligonucleotides encoding peptides of interest, this protocol outlines a fully in vitro and massively parallel procedure for synthesizing the encoded peptides and covalently linking each to a corresponding cDNA tag. The resulting libraries of peptide/DNA conjugates can be used for highly multiplexed assays that leverage high-throughput sequencing to profile the binding or enzymatic specificities of proteins of interest. Here, we describe the implementation of PepSeq for fast and cost-effective epitope-level analysis of antibody reactivity across hundreds of thousands of peptides from <1 µl of serum or plasma input. This protocol includes the design of the DNA oligonucleotide library, synthesis of DNA-barcoded peptide constructs, binding of constructs to sample, preparation for sequencing and data analysis. Implemented in this way, PepSeq can be used for a number of applications, including fine-scale mapping of antibody epitopes and determining a subject's pathogen exposure history. The protocol is divided into two main sections: (i) design and synthesis of DNA-barcoded peptide libraries and (ii) use of libraries for highly multiplexed serology. Once oligonucleotide templates are in hand, library synthesis takes 1-2 weeks and can provide enough material for hundreds to thousands of assays. Serological assays can be conducted in 96-well plates and generate sequencing data within a further ~4 d. A suite of software tools, including the PepSIRF package, are made available to facilitate the design of PepSeq libraries and analysis of assay data.
© 2022. Springer Nature Limited.
Conflict of interest statement
Ethics declarations
Competing interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
