

Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor
- PMID: 36388765
- PMCID: PMC9646490
- DOI: 10.1016/j.xgen.2022.100179
Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor
Abstract
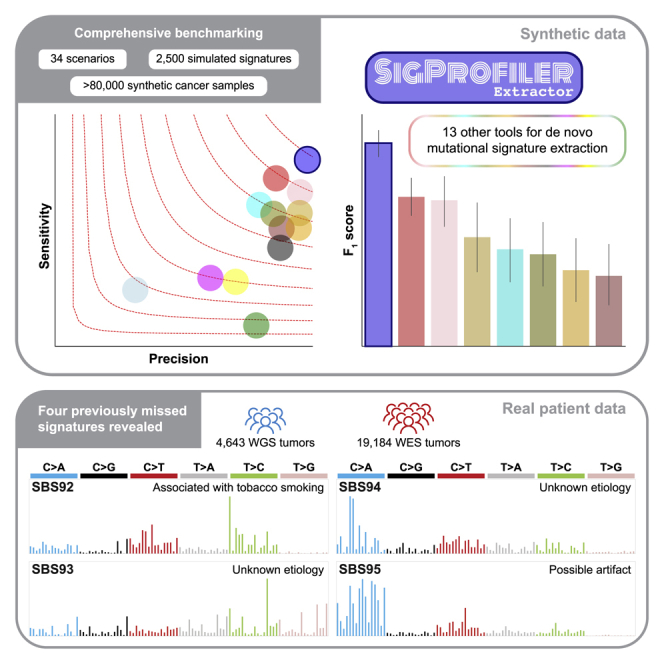
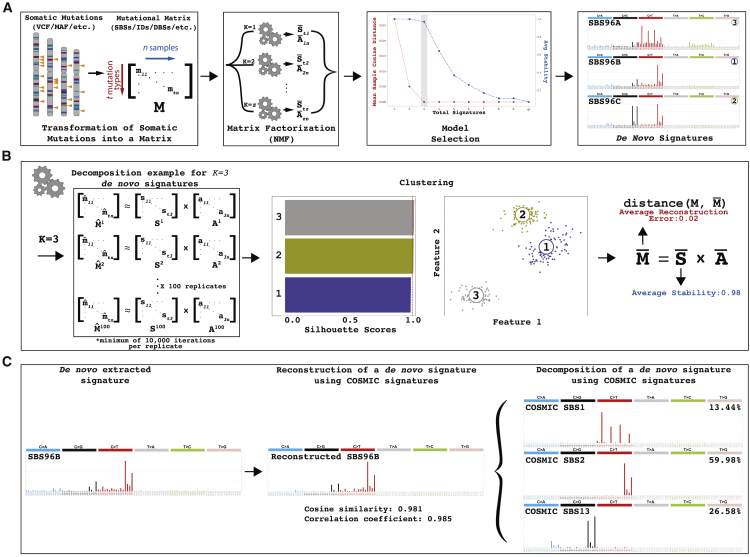
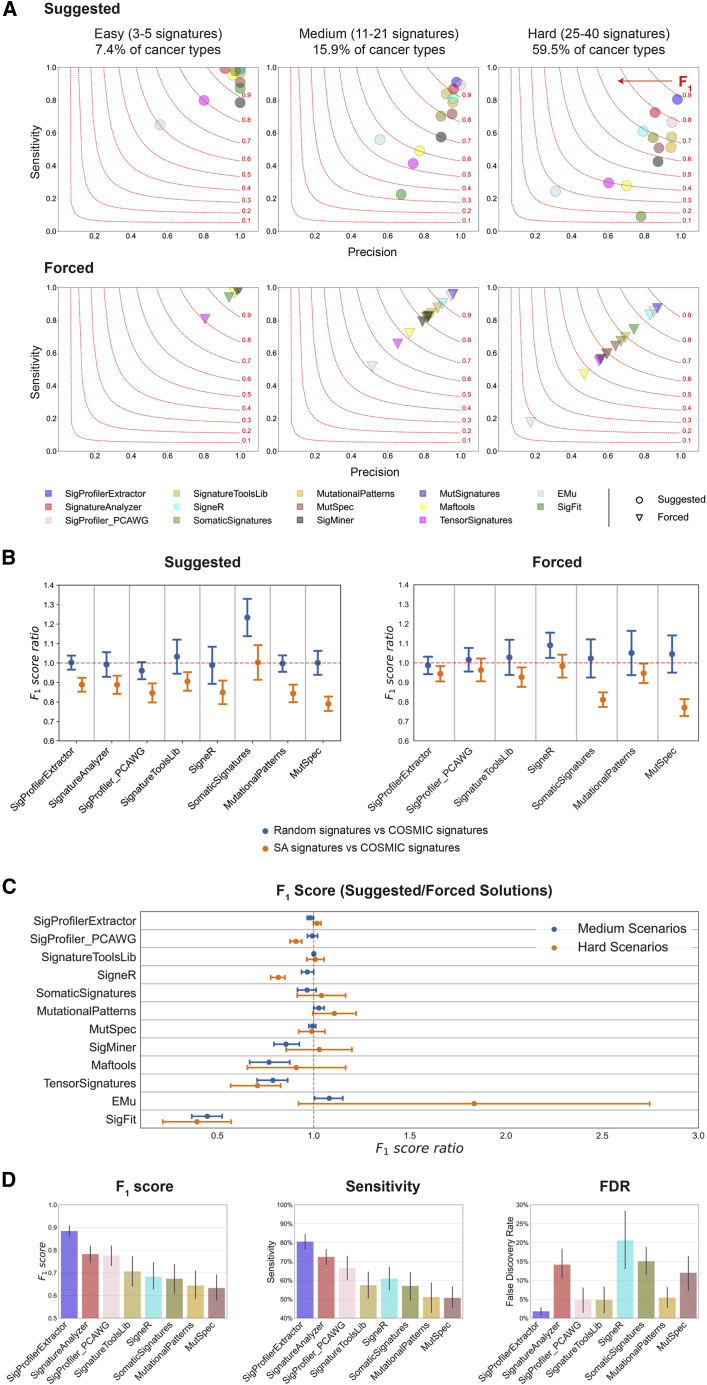
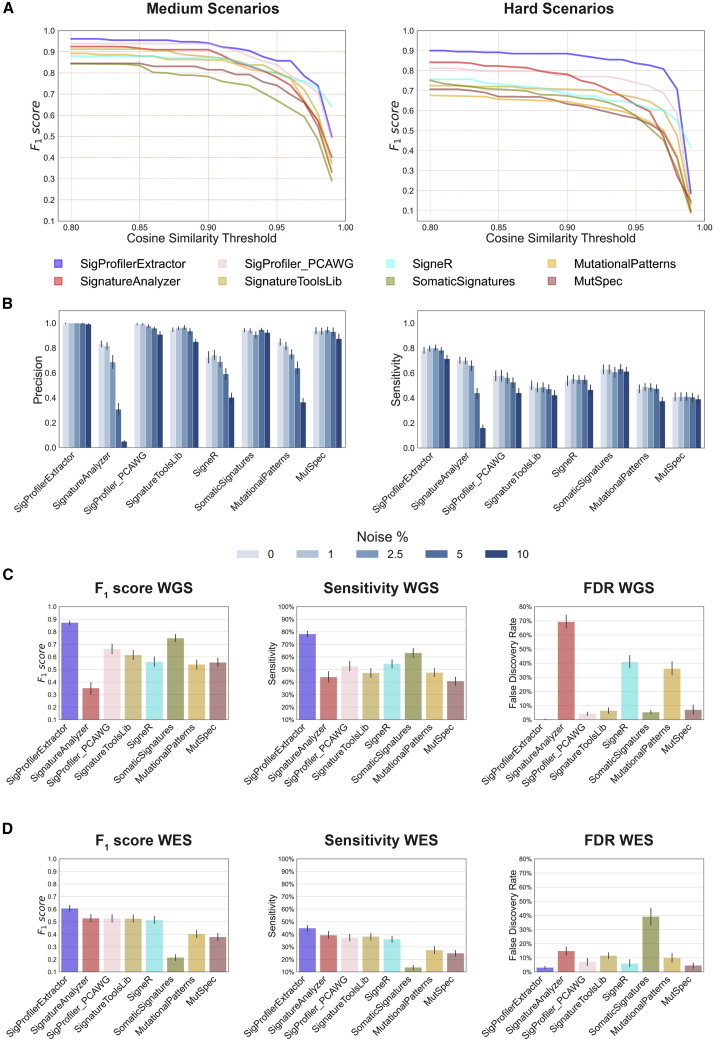
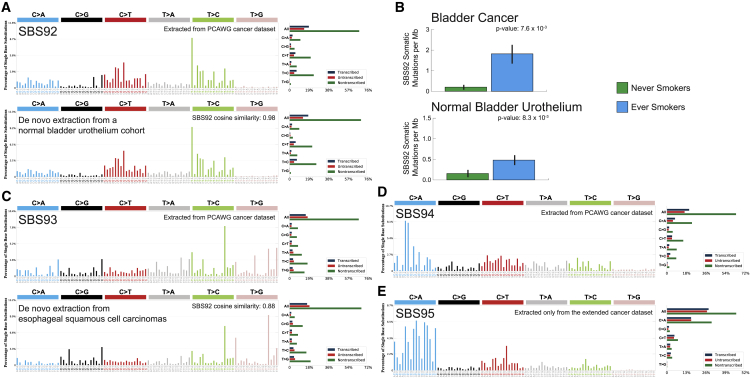
Mutational signature analysis is commonly performed in cancer genomic studies. Here, we present SigProfilerExtractor, an automated tool for de novo extraction of mutational signatures, and benchmark it against another 13 bioinformatics tools by using 34 scenarios encompassing 2,500 simulated signatures found in 60,000 synthetic genomes and 20,000 synthetic exomes. For simulations with 5% noise, reflecting high-quality datasets, SigProfilerExtractor outperforms other approaches by elucidating between 20% and 50% more true-positive signatures while yielding 5-fold less false-positive signatures. Applying SigProfilerExtractor to 4,643 whole-genome- and 19,184 whole-exome-sequenced cancers reveals four novel signatures. Two of the signatures are confirmed in independent cohorts, and one of these signatures is associated with tobacco smoking. In summary, this report provides a reference tool for analysis of mutational signatures, a comprehensive benchmarking of bioinformatics tools for extracting signatures, and several novel mutational signatures, including one putatively attributed to direct tobacco smoking mutagenesis in bladder tissues.
Keywords: cancer genomics; genomics; mutagenesis; mutational signatures.
© 2022 The Author(s).
Conflict of interest statement
M.V. is an employee of NVIDIA corporation. L.B.A. is a compensated consultant and has equity interest in io9, LLC. His spouse is an employee of Biotheranostics, Inc. L.B.A. and B.S.A. are inventors of a US patent 10,776,718. E.N.B. and L.B.A. declare US provisional applications with serial numbers 63/289,601 and 63/269,033. L.B.A. and A.A. declare US provisional patent applications with serial numbers 63/366,392 and 63/367,846. All other authors declare no competing interests.
Figures
References
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
