

HSF1 and Its Role in Huntington's Disease Pathology
- PMID: 36396925
- PMCID: PMC12001818
- DOI: 10.1007/5584_2022_742
HSF1 and Its Role in Huntington's Disease Pathology
Abstract
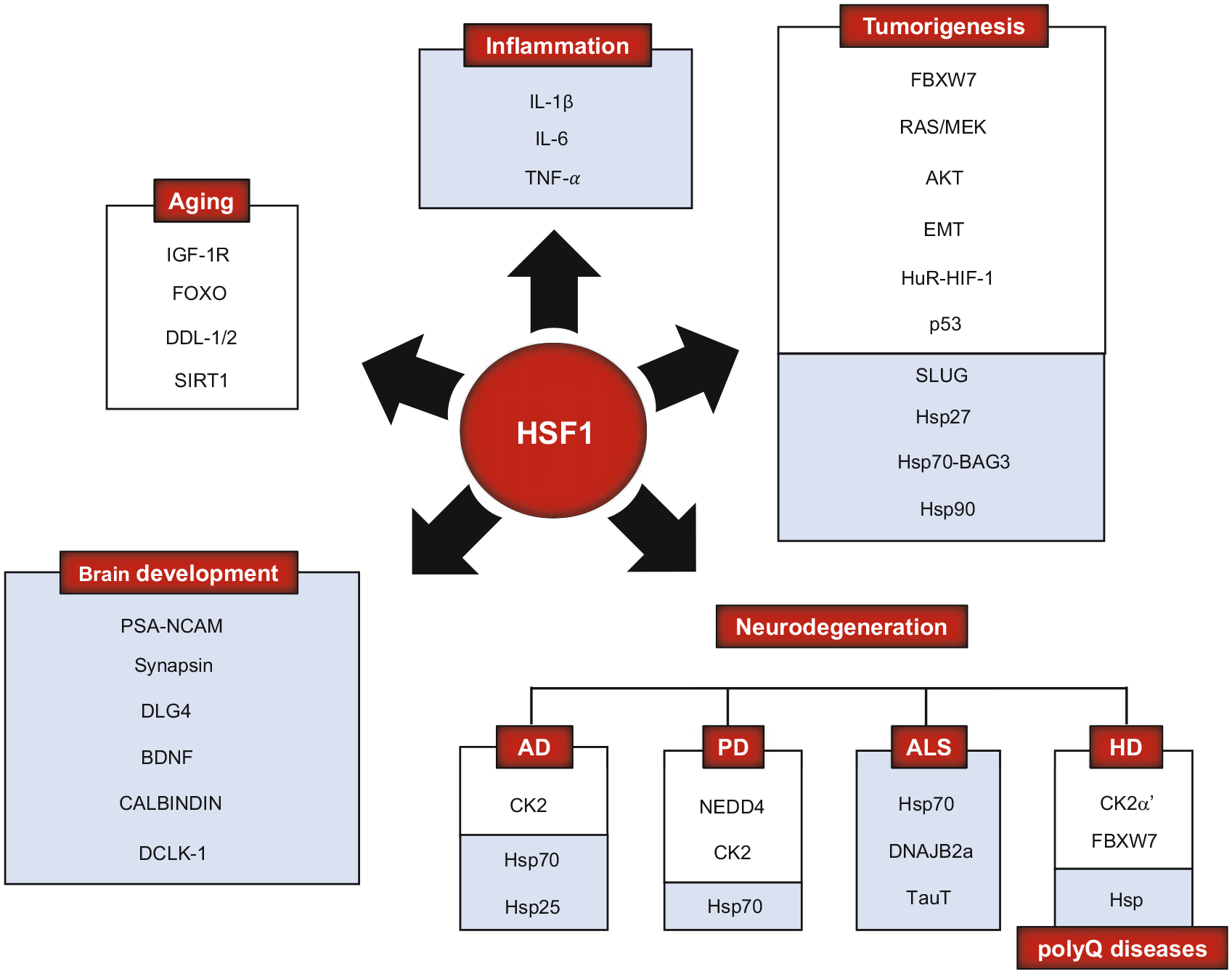
Purpose of review: Heat shock factor 1 (HSF1) is the master transcriptional regulator of the heat shock response (HSR) in mammalian cells and is a critical element in maintaining protein homeostasis. HSF1 functions at the center of many physiological processes like embryogenesis, metabolism, immune response, aging, cancer, and neurodegeneration. However, the mechanisms that allow HSF1 to control these different biological and pathophysiological processes are not fully understood. This review focuses on Huntington's disease (HD), a neurodegenerative disease characterized by severe protein aggregation of the huntingtin (HTT) protein. The aggregation of HTT, in turn, leads to a halt in the function of HSF1. Understanding the pathways that regulate HSF1 in different contexts like HD may hold the key to understanding the pathomechanisms underlying other proteinopathies. We provide the most current information on HSF1 structure, function, and regulation, emphasizing HD, and discussing its potential as a biological target for therapy.
Data sources: We performed PubMed search to find established and recent reports in HSF1, heat shock proteins (Hsp), HD, Hsp inhibitors, HSF1 activators, and HSF1 in aging, inflammation, cancer, brain development, mitochondria, synaptic plasticity, polyglutamine (polyQ) diseases, and HD.
Study selections: Research and review articles that described the mechanisms of action of HSF1 were selected based on terms used in PubMed search.
Results: HSF1 plays a crucial role in the progression of HD and other protein-misfolding related neurodegenerative diseases. Different animal models of HD, as well as postmortem brains of patients with HD, reveal a connection between the levels of HSF1 and HSF1 dysfunction to mutant HTT (mHTT)-induced toxicity and protein aggregation, dysregulation of the ubiquitin-proteasome system (UPS), oxidative stress, mitochondrial dysfunction, and disruption of the structural and functional integrity of synaptic connections, which eventually leads to neuronal loss. These features are shared with other neurodegenerative diseases (NDs). Currently, several inhibitors against negative regulators of HSF1, as well as HSF1 activators, are developed and hold promise to prevent neurodegeneration in HD and other NDs.
Conclusion: Understanding the role of HSF1 during protein aggregation and neurodegeneration in HD may help to develop therapeutic strategies that could be effective across different NDs.
Keywords: Aggregation; Heat shock factor (HSF1); Heat shock proteins (Hsp); Huntington’s diseases (HD); Mitochondria.
© 2022. The Author(s), under exclusive license to Springer Nature Switzerland AG.
Conflict of interest statement
Figures







References
-
- Acebron SP, Martin I, del Castillo U, Moro F, Muga A (2009) DnaK-mediated association of ClpB to protein aggregates. A bichaperone network at the aggregate surface. FEBS Lett 583:2991–2996 - PubMed
-
- Adachi H, Katsuno M, Minamiyama M et al. (2005) Wide-spread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 128:659–670 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
