

PPARγ lipodystrophy mutants reveal intermolecular interactions required for enhancer activation
- PMID: 36402763
- PMCID: PMC9675755
- DOI: 10.1038/s41467-022-34766-9
PPARγ lipodystrophy mutants reveal intermolecular interactions required for enhancer activation
Abstract
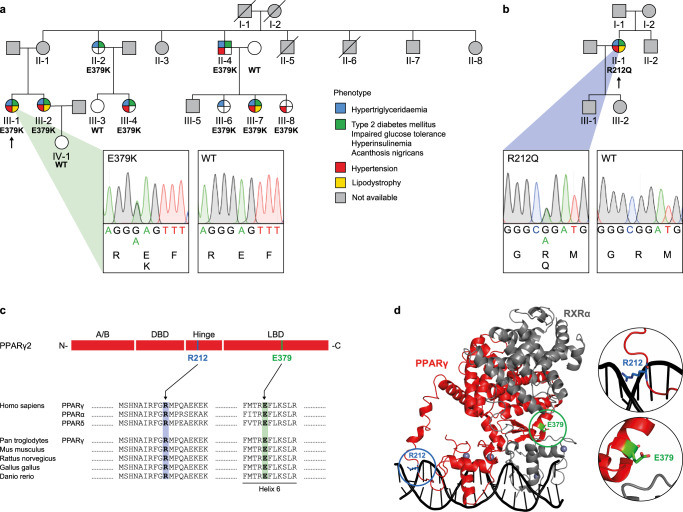
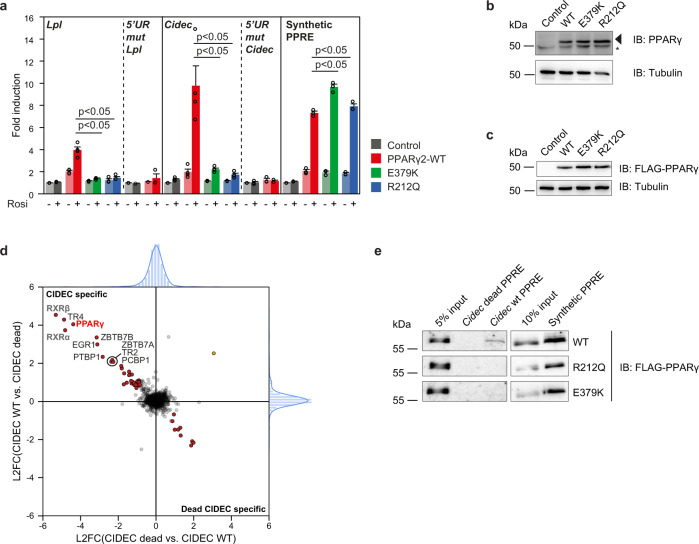
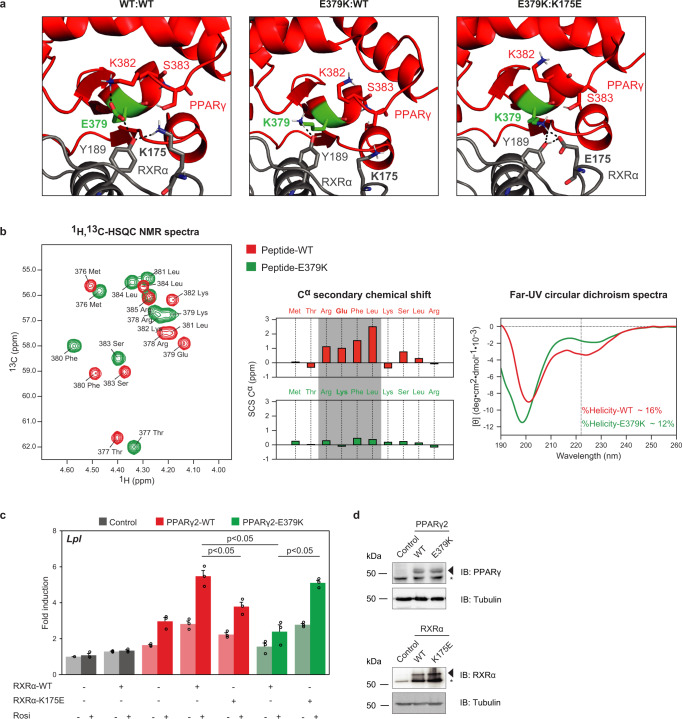
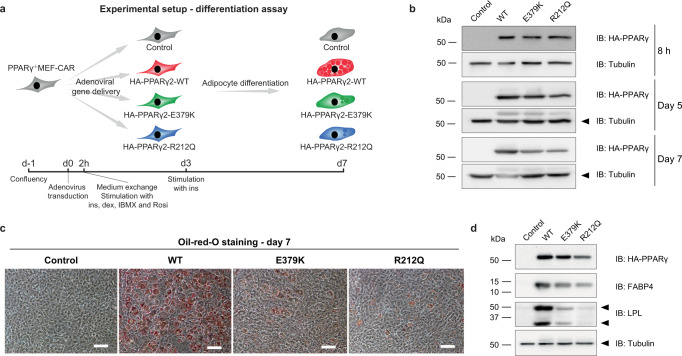
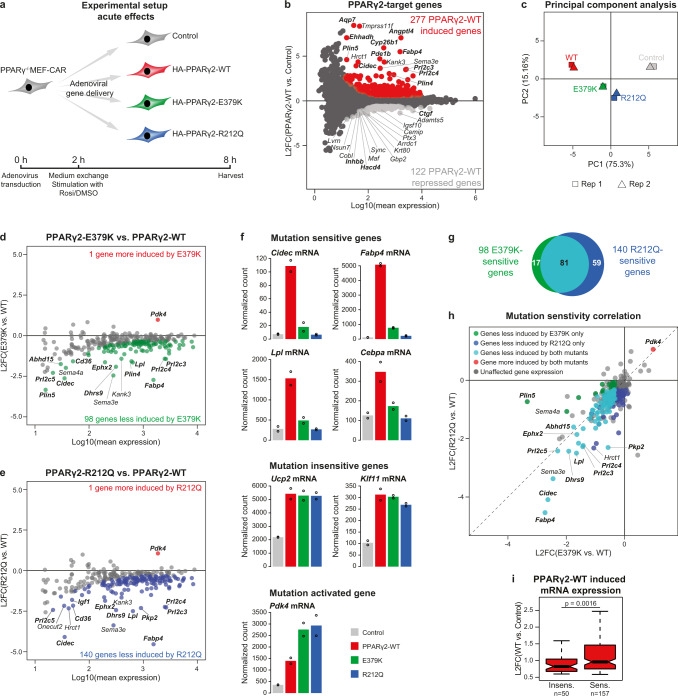
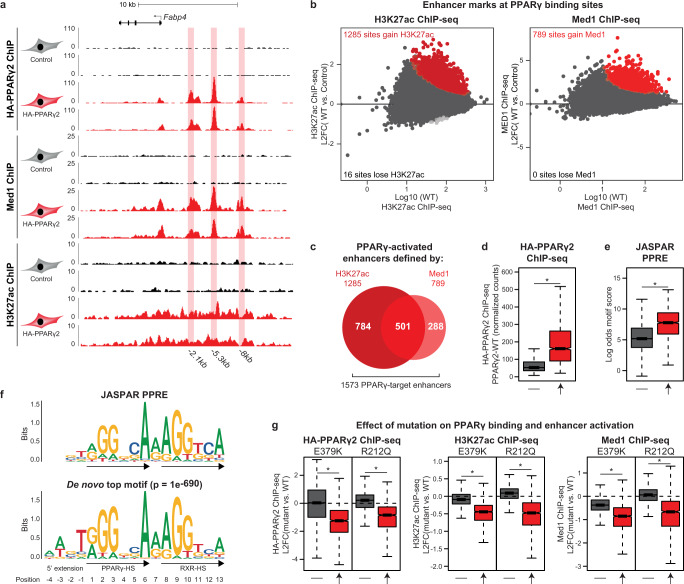
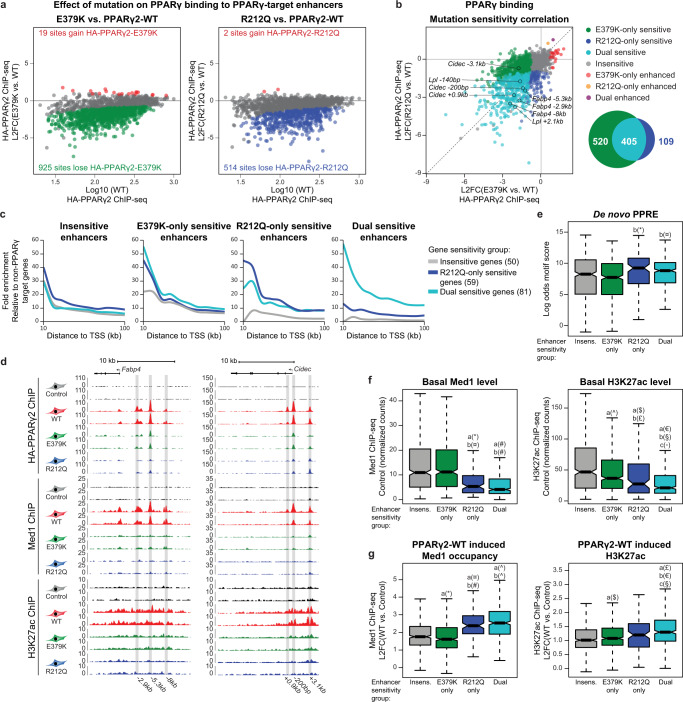
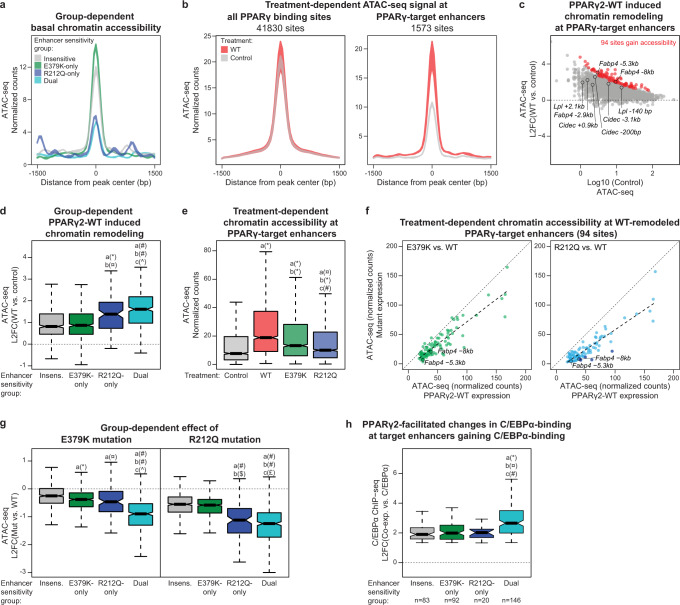
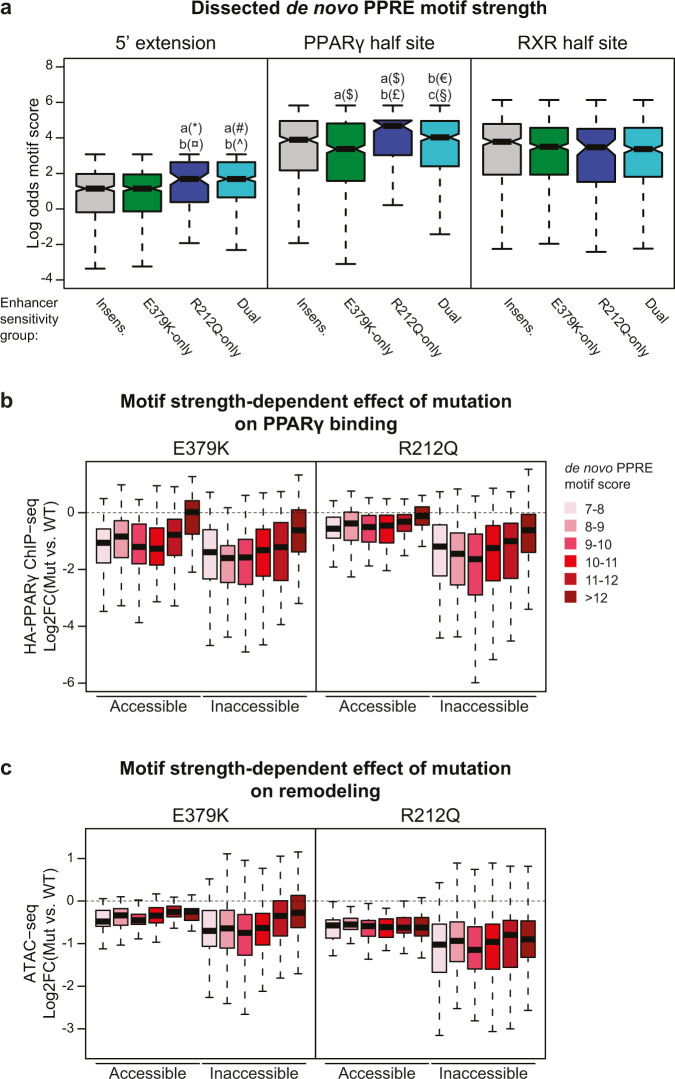
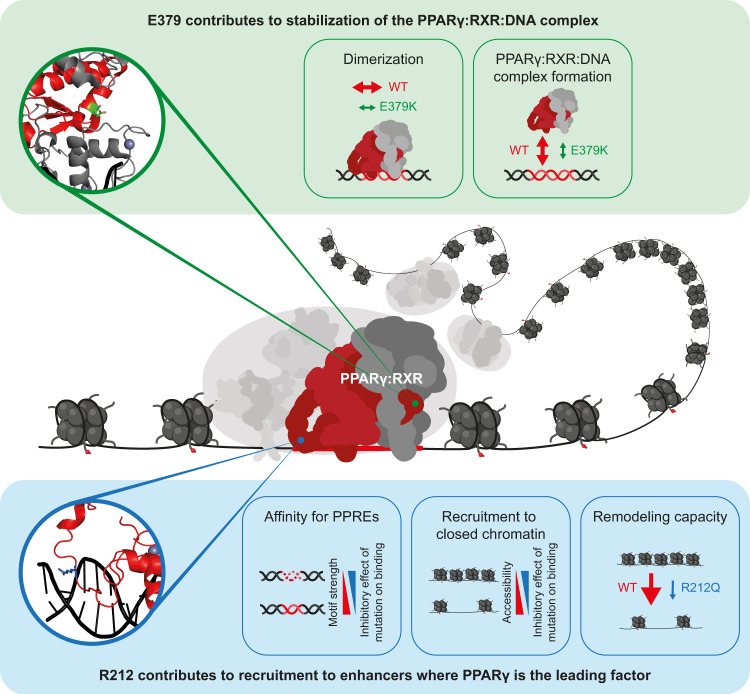
Peroxisome proliferator-activated receptor γ (PPARγ) is the master regulator of adipocyte differentiation, and mutations that interfere with its function cause lipodystrophy. PPARγ is a highly modular protein, and structural studies indicate that PPARγ domains engage in several intra- and inter-molecular interactions. How these interactions modulate PPARγ's ability to activate target genes in a cellular context is currently poorly understood. Here we take advantage of two previously uncharacterized lipodystrophy mutations, R212Q and E379K, that are predicted to interfere with the interaction of the hinge of PPARγ with DNA and with the interaction of PPARγ ligand binding domain (LBD) with the DNA-binding domain (DBD) of the retinoid X receptor, respectively. Using biochemical and genome-wide approaches we show that these mutations impair PPARγ function on an overlapping subset of target enhancers. The hinge region-DNA interaction appears mostly important for binding and remodelling of target enhancers in inaccessible chromatin, whereas the PPARγ-LBD:RXR-DBD interface stabilizes the PPARγ:RXR:DNA ternary complex. Our data demonstrate how in-depth analyses of lipodystrophy mutants can unravel molecular mechanisms of PPARγ function.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- IJpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J. Biol. Chem. 1997;272:20108–20117. doi: 10.1074/jbc.272.32.20108. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
