

Pharmacological inhibition of HDAC6 improves muscle phenotypes in dystrophin-deficient mice by downregulating TGF-β via Smad3 acetylation
- PMID: 36402791
- PMCID: PMC9675748
- DOI: 10.1038/s41467-022-34831-3
Pharmacological inhibition of HDAC6 improves muscle phenotypes in dystrophin-deficient mice by downregulating TGF-β via Smad3 acetylation
Abstract
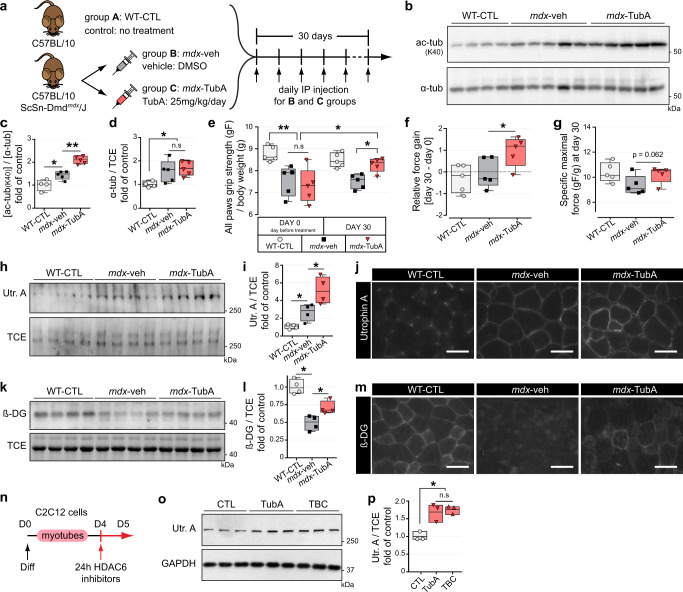
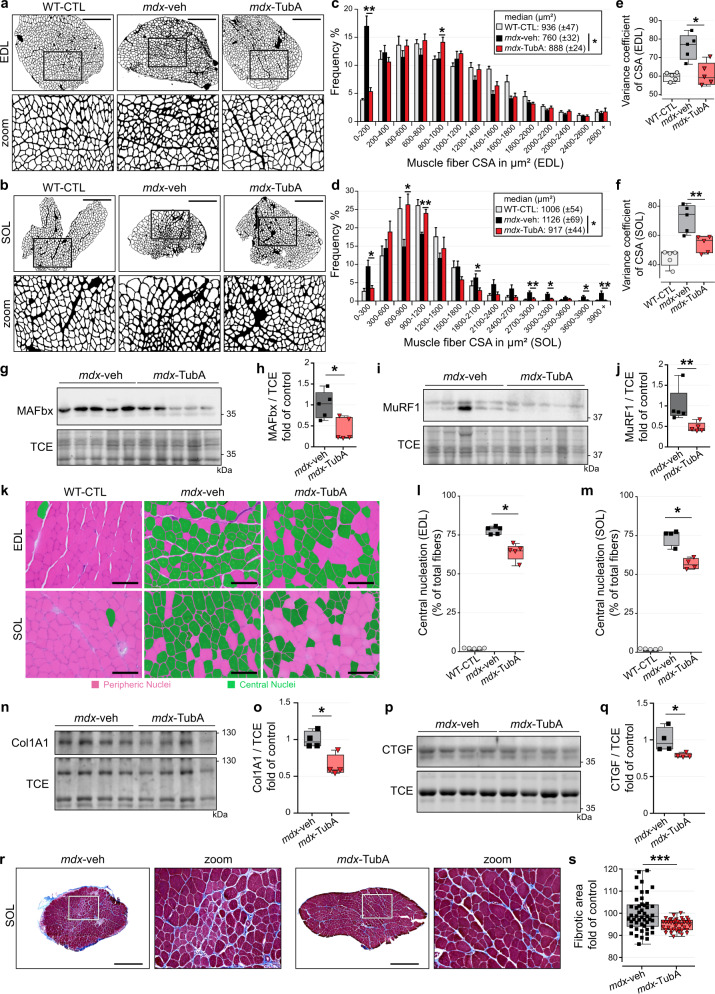
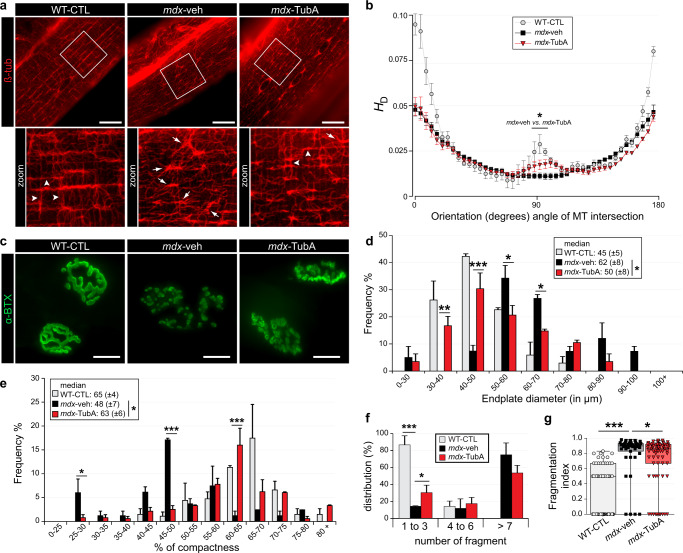
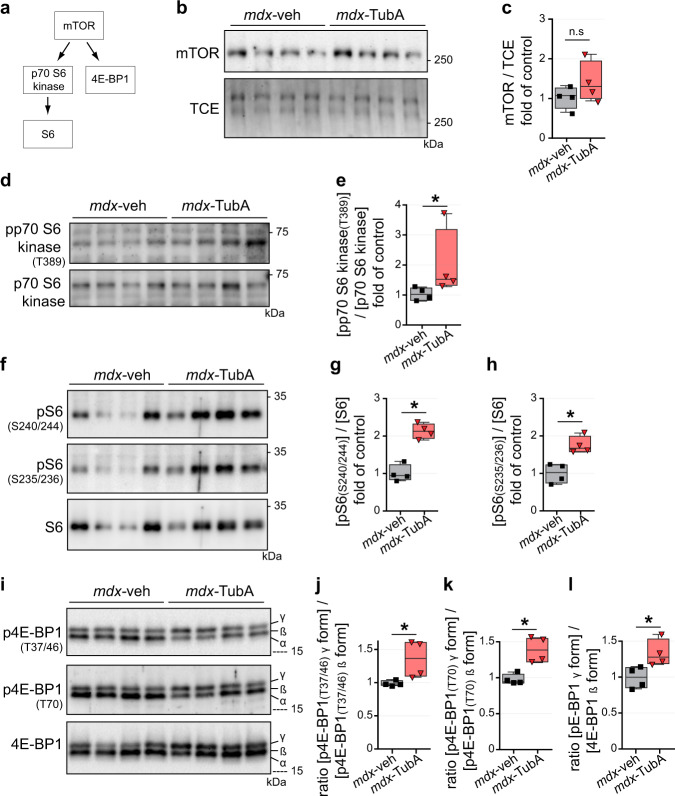
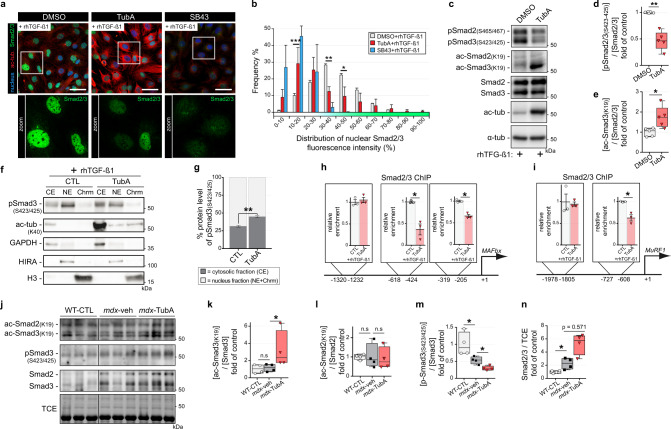
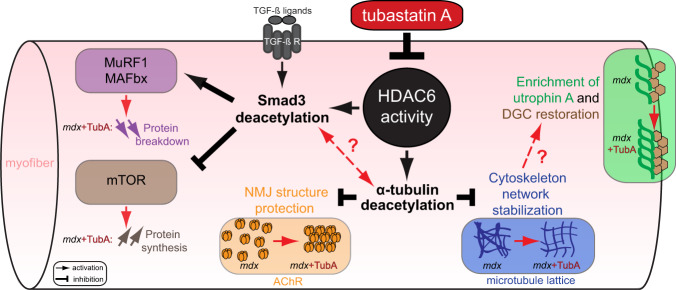
The absence of dystrophin in Duchenne muscular dystrophy disrupts the dystrophin-associated glycoprotein complex resulting in skeletal muscle fiber fragility and atrophy, associated with fibrosis as well as microtubule and neuromuscular junction disorganization. The specific, non-conventional cytoplasmic histone deacetylase 6 (HDAC6) was recently shown to regulate acetylcholine receptor distribution and muscle atrophy. Here, we report that administration of the HDAC6 selective inhibitor tubastatin A to the Duchenne muscular dystrophy, mdx mouse model increases muscle strength, improves microtubule, neuromuscular junction, and dystrophin-associated glycoprotein complex organization, and reduces muscle atrophy and fibrosis. Interestingly, we found that the beneficial effects of HDAC6 inhibition involve the downregulation of transforming growth factor beta signaling. By increasing Smad3 acetylation in the cytoplasm, HDAC6 inhibition reduces Smad2/3 phosphorylation, nuclear translocation, and transcriptional activity. These findings provide in vivo evidence that Smad3 is a new target of HDAC6 and implicate HDAC6 as a potential therapeutic target in Duchenne muscular dystrophy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
