

Mito-SiPE is a sequence-independent and PCR-free mtDNA enrichment method for accurate ultra-deep mitochondrial sequencing
- PMID: 36402890
- PMCID: PMC9675811
- DOI: 10.1038/s42003-022-04182-2
Mito-SiPE is a sequence-independent and PCR-free mtDNA enrichment method for accurate ultra-deep mitochondrial sequencing
Abstract
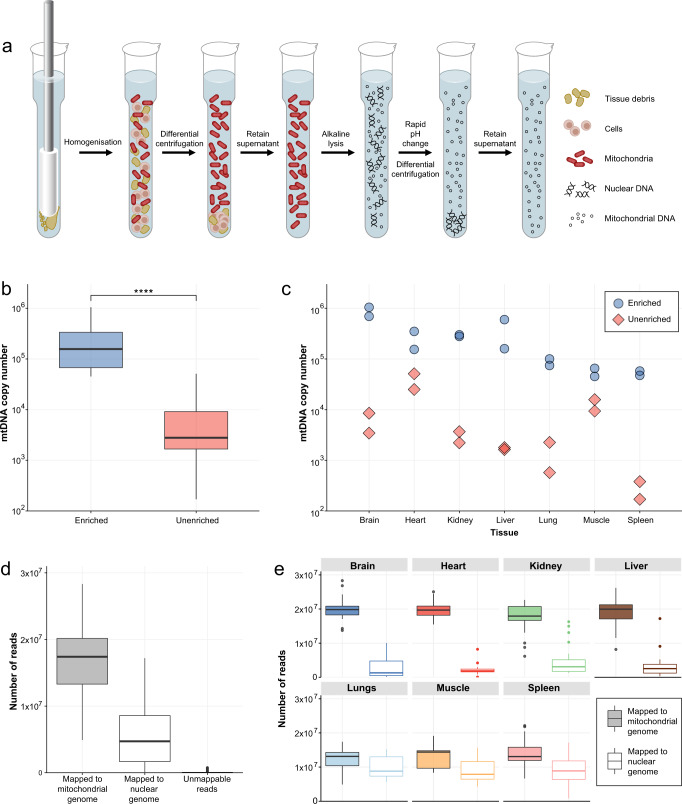
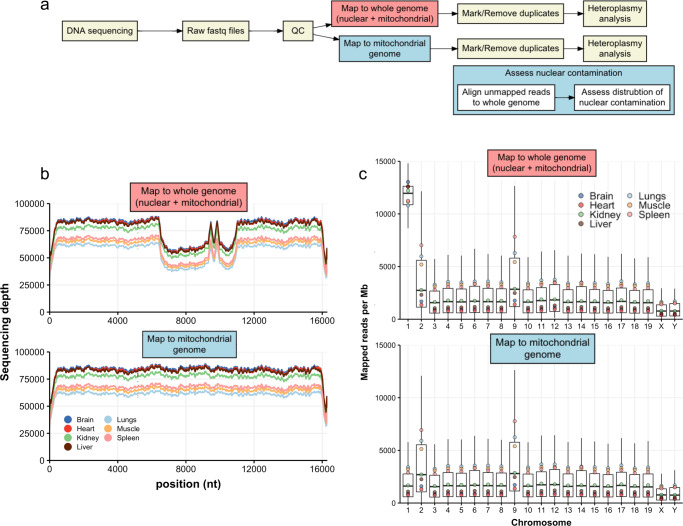
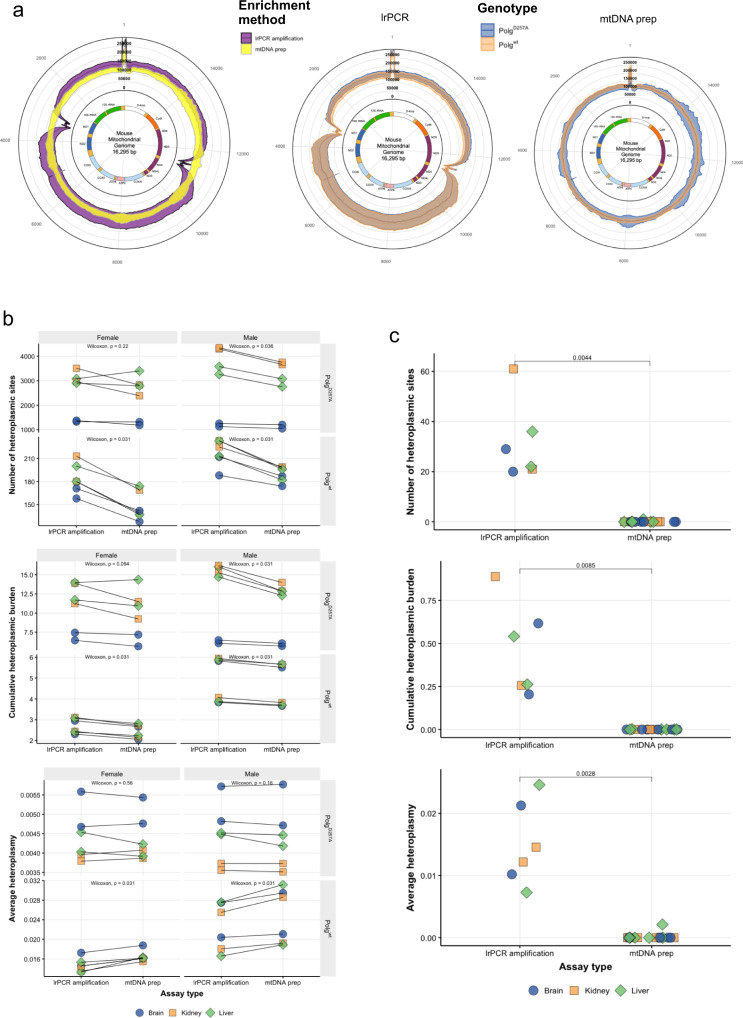
The analysis of somatic variation in the mitochondrial genome requires deep sequencing of mitochondrial DNA. This is ordinarily achieved by selective enrichment methods, such as PCR amplification or probe hybridization. These methods can introduce bias and are prone to contamination by nuclear-mitochondrial sequences (NUMTs), elements that can introduce artefacts into heteroplasmy analysis. We isolated intact mitochondria using differential centrifugation and alkaline lysis and subjected purified mitochondrial DNA to a sequence-independent and PCR-free method to obtain ultra-deep (>80,000X) sequencing coverage of the mitochondrial genome. This methodology avoids false-heteroplasmy calls that occur when long-range PCR amplification is used for mitochondrial DNA enrichment. Previously published methods employing mitochondrial DNA purification did not measure mitochondrial DNA enrichment or utilise high coverage short-read sequencing. Here, we describe a protocol that yields mitochondrial DNA and have quantified the increased level of mitochondrial DNA post-enrichment in 7 different mouse tissues. This method will enable researchers to identify changes in low frequency heteroplasmy without introducing PCR biases or NUMT contamination that are incorrectly identified as heteroplasmy when long-range PCR is used.
© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Sequencing and characterizing human mitochondrial genomes in the biobank-based genomic research paradigm.Sci China Life Sci. 2025 Jun;68(6):1610-1625. doi: 10.1007/s11427-024-2736-7. Epub 2025 Jan 21. Sci China Life Sci. 2025. PMID: 39843848 Review.
-
PCR-Free Enrichment of Mitochondrial DNA from Human Blood and Cell Lines for High Quality Next-Generation DNA Sequencing.PLoS One. 2015 Oct 21;10(10):e0139253. doi: 10.1371/journal.pone.0139253. eCollection 2015. PLoS One. 2015. PMID: 26488301 Free PMC article.
-
Mitochondrial DNA enrichment reduced NUMT contamination in porcine NGS analyses.Brief Bioinform. 2020 Jul 15;21(4):1368-1377. doi: 10.1093/bib/bbz060. Brief Bioinform. 2020. PMID: 31204429
-
Sensitivity of mitochondrial DNA heteroplasmy detection using Next Generation Sequencing.Mitochondrion. 2020 Jan;50:88-93. doi: 10.1016/j.mito.2019.10.006. Epub 2019 Oct 25. Mitochondrion. 2020. PMID: 31669622
-
Recent Advances in Detecting Mitochondrial DNA Heteroplasmic Variations.Molecules. 2018 Feb 3;23(2):323. doi: 10.3390/molecules23020323. Molecules. 2018. PMID: 29401641 Free PMC article. Review.
Cited by
-
The Mighty NUMT: Mitochondrial DNA Flexing Its Code in the Nuclear Genome.Biomolecules. 2023 Apr 27;13(5):753. doi: 10.3390/biom13050753. Biomolecules. 2023. PMID: 37238623 Free PMC article. Review.
-
Sequencing and characterizing human mitochondrial genomes in the biobank-based genomic research paradigm.Sci China Life Sci. 2025 Jun;68(6):1610-1625. doi: 10.1007/s11427-024-2736-7. Epub 2025 Jan 21. Sci China Life Sci. 2025. PMID: 39843848 Review.
-
Harnessing tissue-derived mitochondria-rich extracellular vesicles (Ti-mitoEVs) to boost mitochondrial biogenesis for regenerative medicine.Sci Adv. 2025 Jul 18;11(29):eadt1318. doi: 10.1126/sciadv.adt1318. Epub 2025 Jul 16. Sci Adv. 2025. PMID: 40668934 Free PMC article.
-
Deeper insight into ferroptosis: association with Alzheimer's, Parkinson's disease, and brain tumors and their possible treatment by nanomaterials induced ferroptosis.Redox Rep. 2023 Dec;28(1):2269331. doi: 10.1080/13510002.2023.2269331. Epub 2023 Nov 27. Redox Rep. 2023. PMID: 38010378 Free PMC article. Review.
-
A PCR-independent approach for mtDNA enrichment and next-generation sequencing: comprehensive evaluation and clinical application.J Transl Med. 2024 Apr 25;22(1):386. doi: 10.1186/s12967-024-05213-8. J Transl Med. 2024. PMID: 38664838 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
