

Fructose impairs fat oxidation: Implications for the mechanism of western diet-induced NAFLD
- PMID: 36403701
- PMCID: PMC11042502
- DOI: 10.1016/j.jnutbio.2022.109224
Fructose impairs fat oxidation: Implications for the mechanism of western diet-induced NAFLD
Abstract
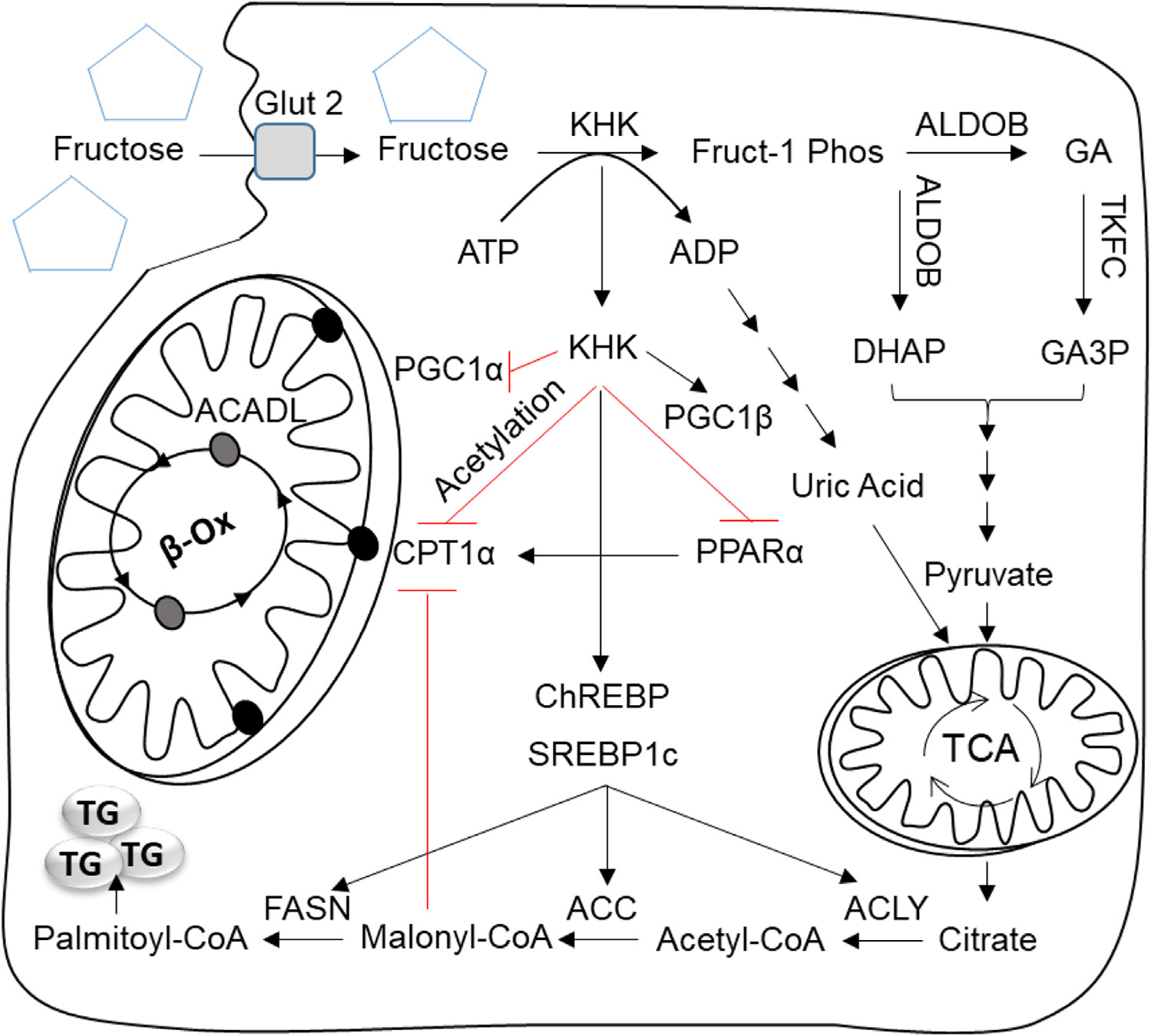
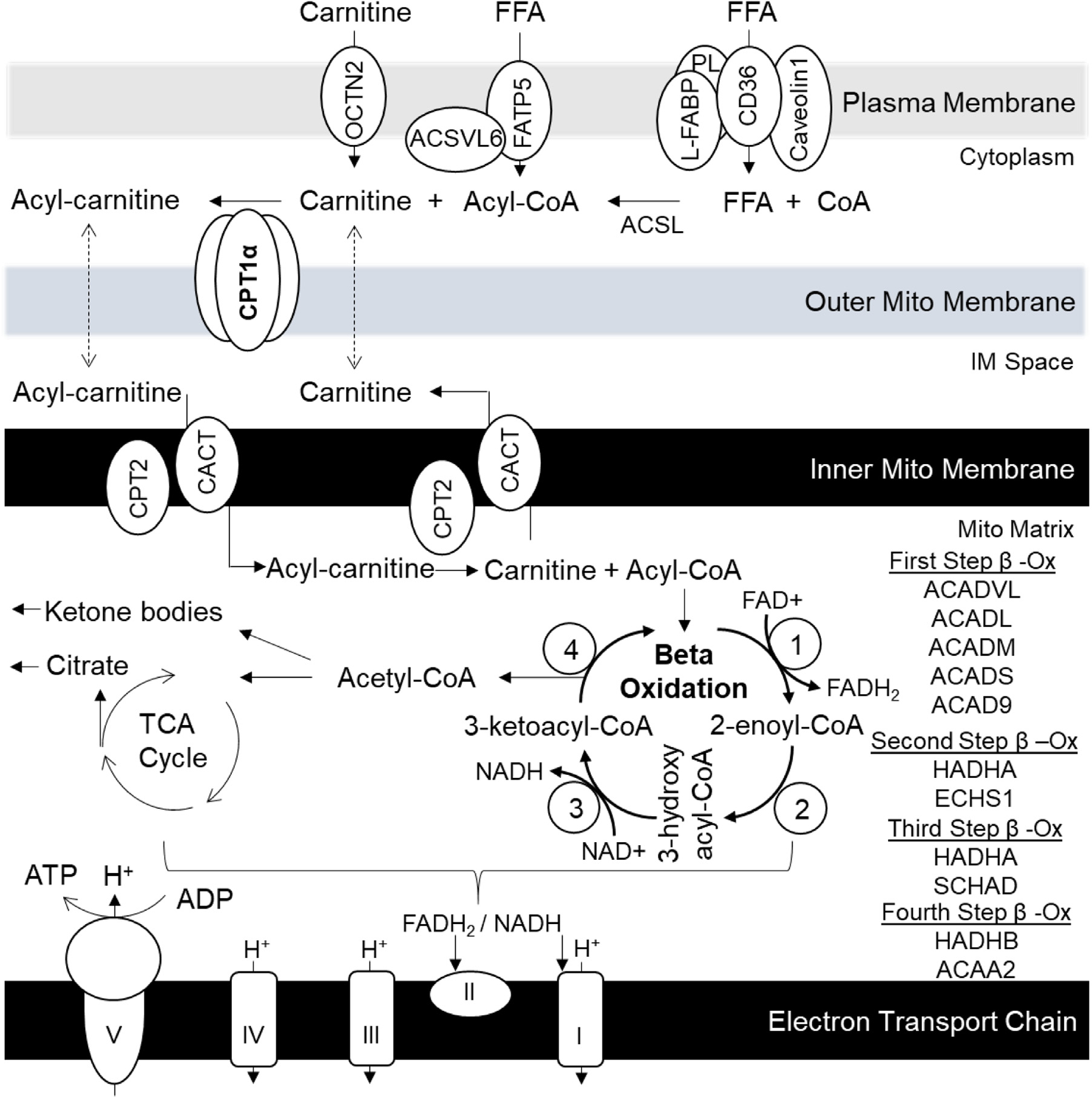
Increased fructose intake from sugar-sweetened beverages and highly processed sweets is a well-recognized risk factor for the development of obesity and its complications. Fructose strongly supports lipogenesis on a normal chow diet by providing both, a substrate for lipid synthesis and activation of lipogenic transcription factors. However, the negative health consequences of dietary sugar are best observed with the concomitant intake of a HFD. Indeed, the most commonly used obesogenic research diets, such as "Western diet", contain both fructose and a high amount of fat. In spite of its common use, how the combined intake of fructose and fat synergistically supports development of metabolic complications is not fully elucidated. Here we present the preponderance of evidence that fructose consumption decreases oxidation of dietary fat in human and animal studies. We provide a detailed review of the mitochondrial β-oxidation pathway. Fructose affects hepatic activation of fatty acyl-CoAs, decreases acylcarnitine production and impairs the carnitine shuttle. Mechanistically, fructose suppresses transcriptional activity of PPARα and its target CPT1α, the rate limiting enzyme of acylcarnitine production. These effects of fructose may be, in part, mediated by protein acetylation. Acetylation of PGC1α, a co-activator of PPARα and acetylation of CPT1α, in part, account for fructose-impaired acylcarnitine production. Interestingly, metabolic effects of fructose in the liver can be largely overcome by carnitine supplementation. In summary, fructose decreases oxidation of dietary fat in the liver, in part, by impairing acylcarnitine production, offering one explanation for the synergistic effects of these nutrients on the development of metabolic complications, such as NAFLD.
Keywords: Fatty acid oxidation (FAO); Fructose; Non-alcoholic fatty liver disease (NAFLD); Sugar; Western diet.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Competing Interests The authors declare that there are no conflicts of interest.
Figures
References
-
- Bray GA, Popkin BM. Dietary fat intake does affect obesity!. Am J Clin Nutr 1998;68:1157–73. - PubMed
-
- Austin GL, Ogden LG, Hill JO. Trends in carbohydrate, fat, and protein intakes and association with energy intake in normal-weight, overweight, and obese individuals: 1971–2006. Am J Clin Nutr 2011;93:836–43. - PubMed
-
- Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr 2004;79:537–43. - PubMed
-
- Sievenpiper JL, de Souza RJ, Mirrahimi A, Yu ME, Carleton AJ, Beyene J, et al. Effect of fructose on body weight in controlled feeding trials: a systematic review and meta-analysis. Ann Intern Med 2012;156:291–304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
