

Membranous nephropathy: Clearer pathology and mechanisms identify potential strategies for treatment
- PMID: 36405681
- PMCID: PMC9667740
- DOI: 10.3389/fimmu.2022.1036249
Membranous nephropathy: Clearer pathology and mechanisms identify potential strategies for treatment
Abstract
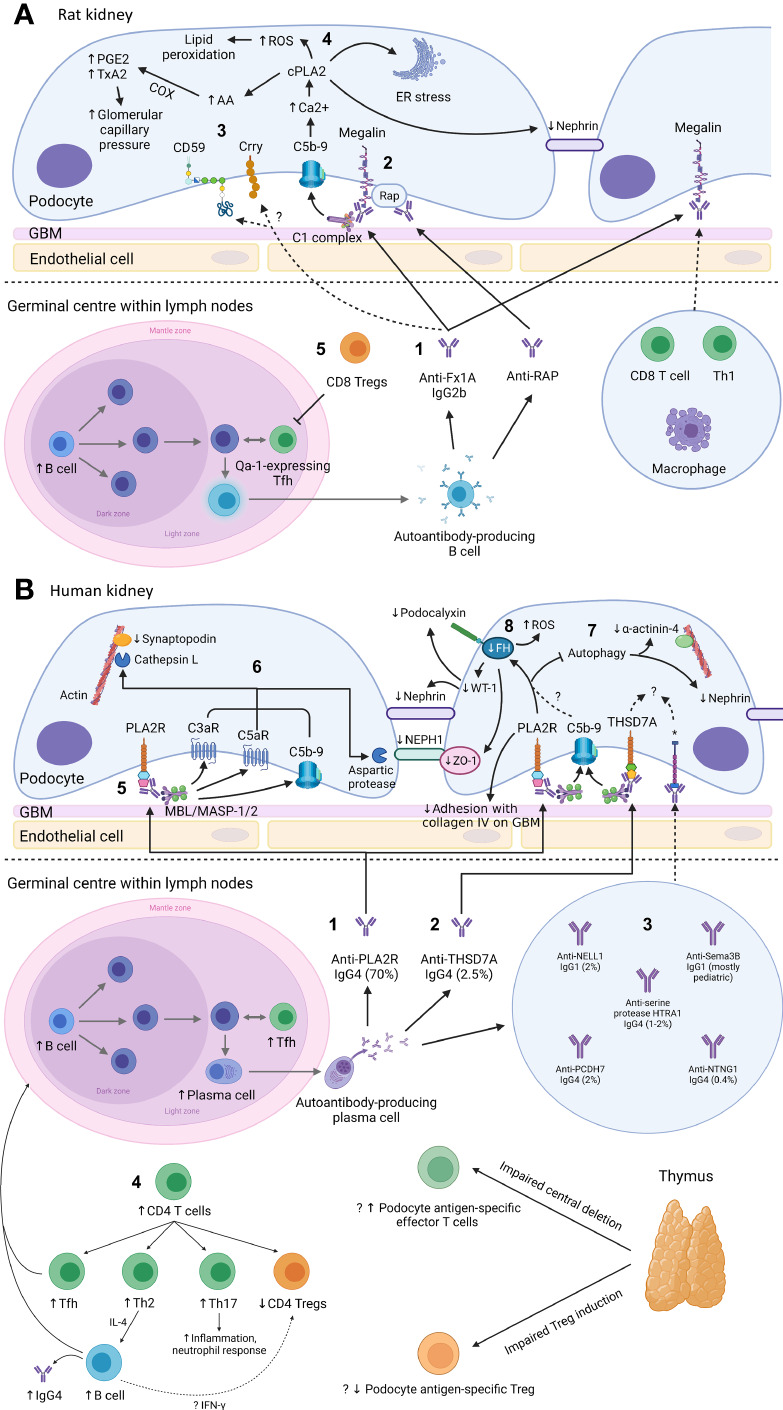
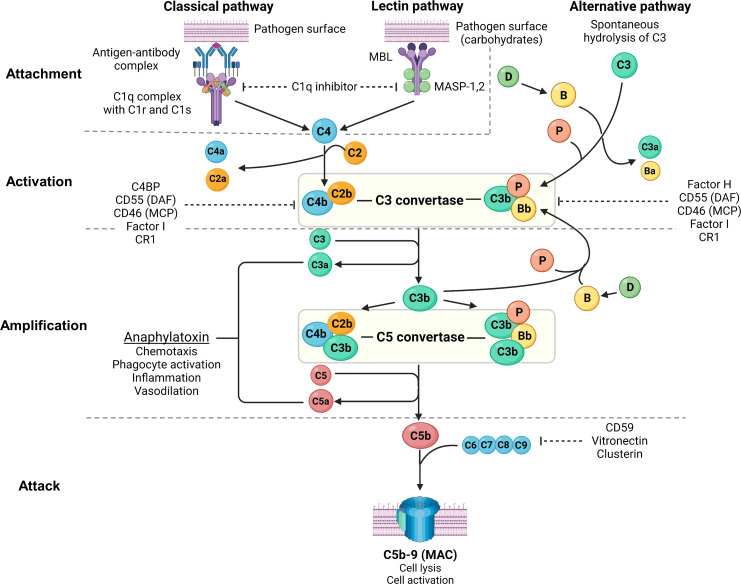
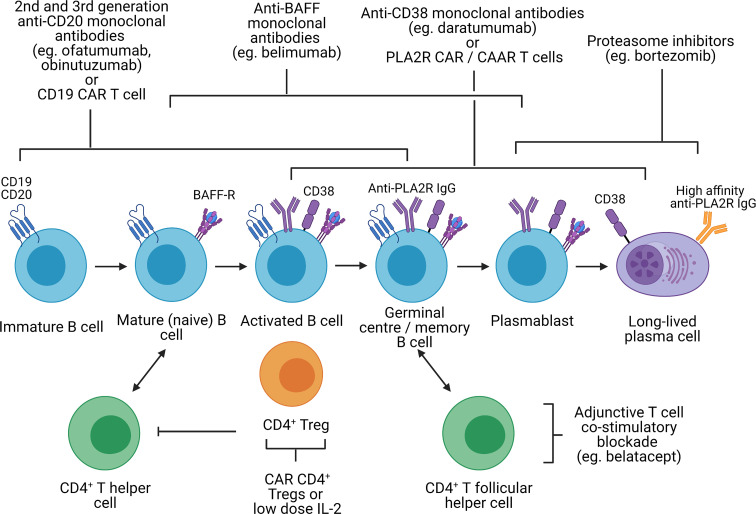
Primary membranous nephropathy (PMN) is one of the common causes of adult-onset nephrotic syndrome and is characterized by autoantibodies against podocyte antigens causing in situ immune complex deposition. Much of our understanding of the disease mechanisms underpinning this kidney-limited autoimmune disease originally came from studies of Heymann nephritis, a rat model of PMN, where autoantibodies against megalin produced a similar disease phenotype though megalin is not implicated in human disease. In PMN, the major target antigen was identified to be M-type phospholipase A2 receptor 1 (PLA2R) in 2009. Further utilization of mass spectrometry on immunoprecipitated glomerular extracts and laser micro dissected glomeruli has allowed the rapid discovery of other antigens (thrombospondin type-1 domain-containing protein 7A, neural epidermal growth factor-like 1 protein, semaphorin 3B, protocadherin 7, high temperature requirement A serine peptidase 1, netrin G1) targeted by autoantibodies in PMN. Despite these major advances in our understanding of the pathophysiology of PMN, treatments remain non-specific, often ineffective, or toxic. In this review, we summarize our current understanding of the immune mechanisms driving PMN from animal models and clinical studies, and the implications on the development of future targeted therapeutic strategies.
Keywords: Heymann nephritis; autoantibody; immunology; membranous nephropathy; podocyte; treatment.
Copyright © 2022 Chung, Wang, Keung, Hu, McCarthy, Wong, Kairaitis, Bose, Harris and Alexander.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources