

Transcriptome and metabolome analyses of Shatian pomelo (Citrus grandis var. Shatinyu Hort) leaves provide insights into the overexpression of the gibberellin-induced gene CcGASA4
- PMID: 36407630
- PMCID: PMC9671072
- DOI: 10.3389/fpls.2022.1022961
Transcriptome and metabolome analyses of Shatian pomelo (Citrus grandis var. Shatinyu Hort) leaves provide insights into the overexpression of the gibberellin-induced gene CcGASA4
Abstract
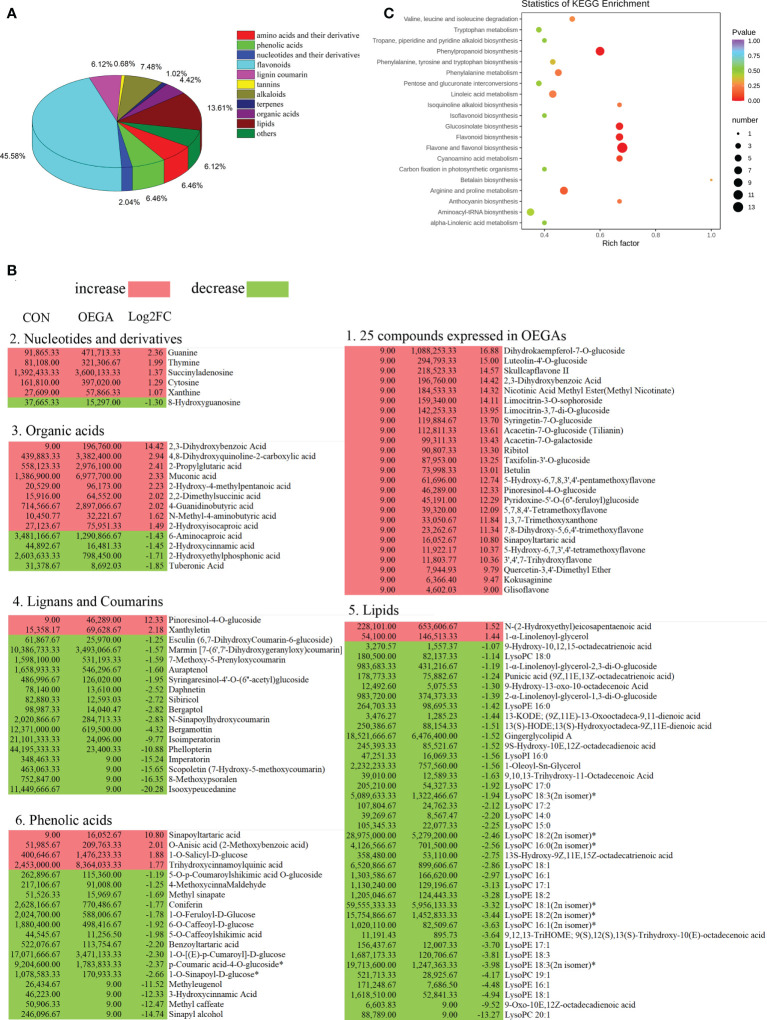
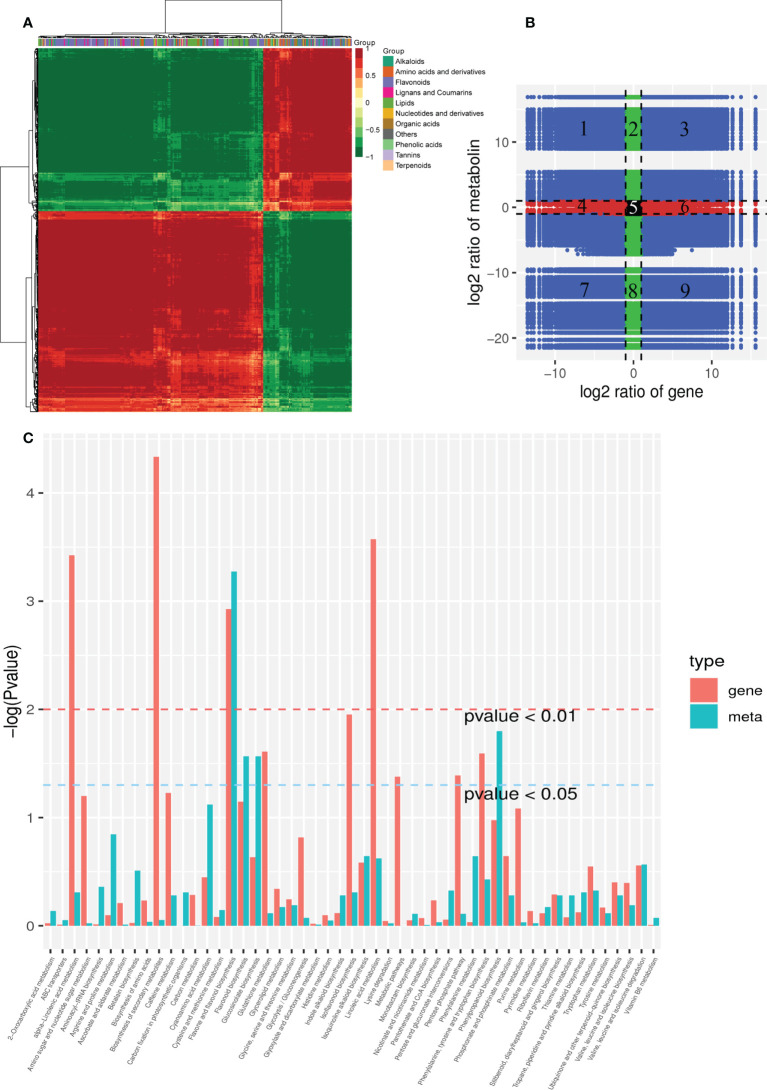
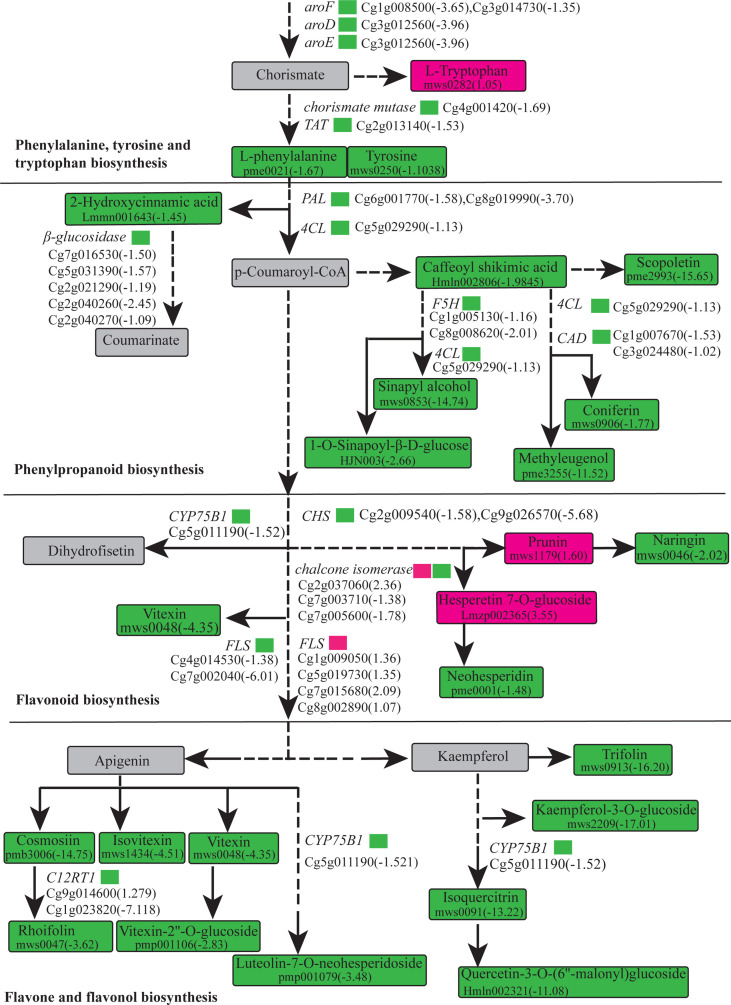
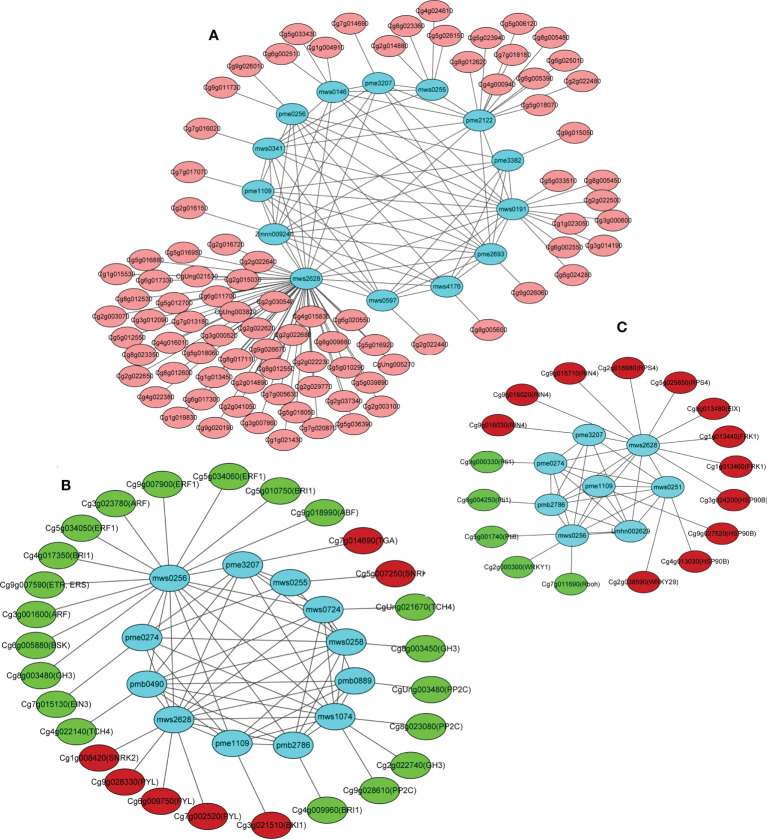
The gibberellic acid (GA)-stimulated Arabidopsis (GASA) gene family is highly specific to plants and plays crucial roles in plant growth and development. CcGASA4 is a member of the GASA gene family in citrus plants; however, the current understanding of its function in citrus is limited. We used CcGASA4-overexpression transgenic citrus (OEGA) and control (CON) plants to study the role of CcGASA4 in Shatian pomelo. The RNA sequencing (RNA-seq) analysis showed that 3,522 genes, including 1,578 upregulated and 1,944 downregulated genes, were significantly differentially expressed in the CON versus OEGA groups. The Gene Ontology enrichment analysis showed that 178 of the differentially-expressed genes (DEGs) were associated with flowers. A Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis showed that the DEGs were enriched in 134 pathways, including "plant-pathogen interaction", "MAPK signaling pathway-plant", "phenylpropane biosynthesis", "plant hormone signal transduction", "phenylalanine, tyrosine and tryptophan biosynthesis", and "flavonoid and flavonol biosynthesis". The most significantly-enriched pathway was "plant-pathogen interaction", in which 203 DEGs were enriched (126 DEGs were upregulated and 78 were downregulated). The metabolome analysis showed that 644 metabolites were detected in the OEGA and CON samples, including 294 differentially-accumulated metabolites (DAMs; 83 upregulated versus 211 downregulated in OEGA compared to CON). The metabolic pathway analysis showed that these DAMs were mainly involved in the metabolic pathways of secondary metabolites, such as phenylpropanoids, phenylalanine, flavone, and flavonol biosynthesis. Thirteen flavonoids and isoflavones were identified as DAMs in OEGA and CON. We also discovered 25 OEGA-specific accumulated metabolites and found 10 that were associated with disease resistance. CcGASA4 may therefore play a functional role in activating the expression of MAPK signaling transduction pathway and disease resistance genes, inhibiting the expression of auxin- and ethylene-related genes, and activating or inhibiting the expression of brassinosteroid biosynthesis- and abscisic acid-related genes. CcGASA4 may also play a role in regulating the composition and abundance of flavonoids, isoflavones, amino acids, purines, and phenolic compounds. This study provides new insights into the molecular mechanisms of action of CcGASA4 in citrus plants.
Keywords: CcGASA4; Citrus; gibberellin; hormone; metabolome; plant–pathogen interaction; transcriptome.
Copyright © 2022 Wu, Liu, Chen, Jiang, Gong and Zhong.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
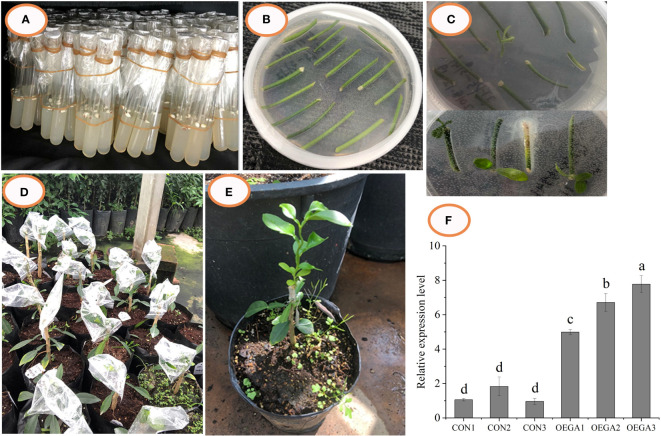
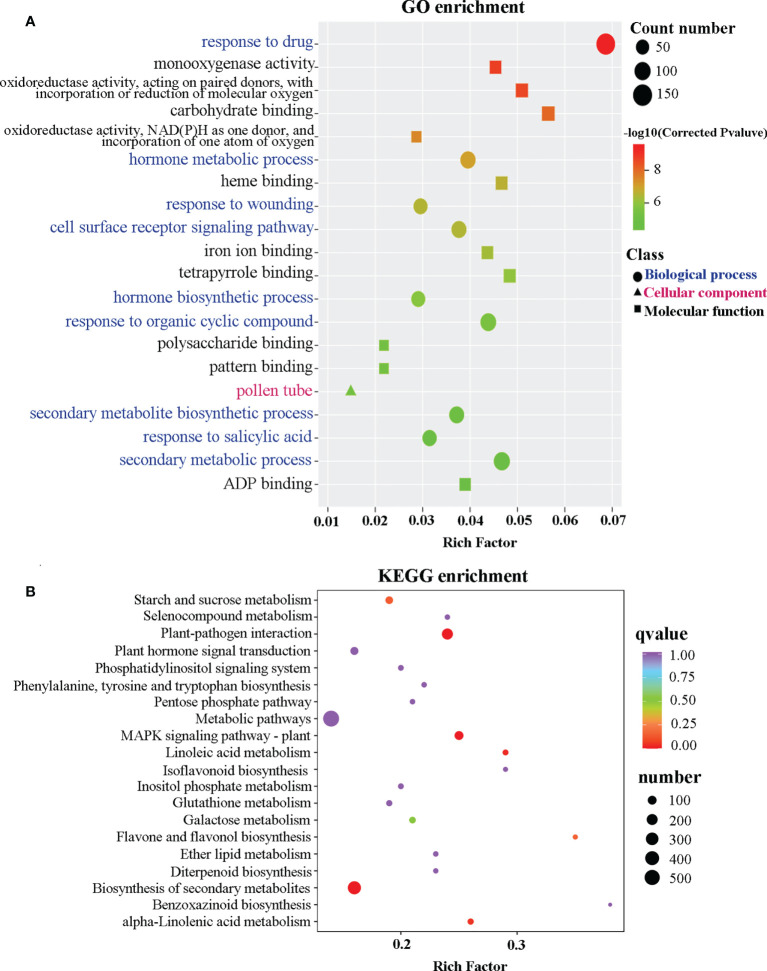
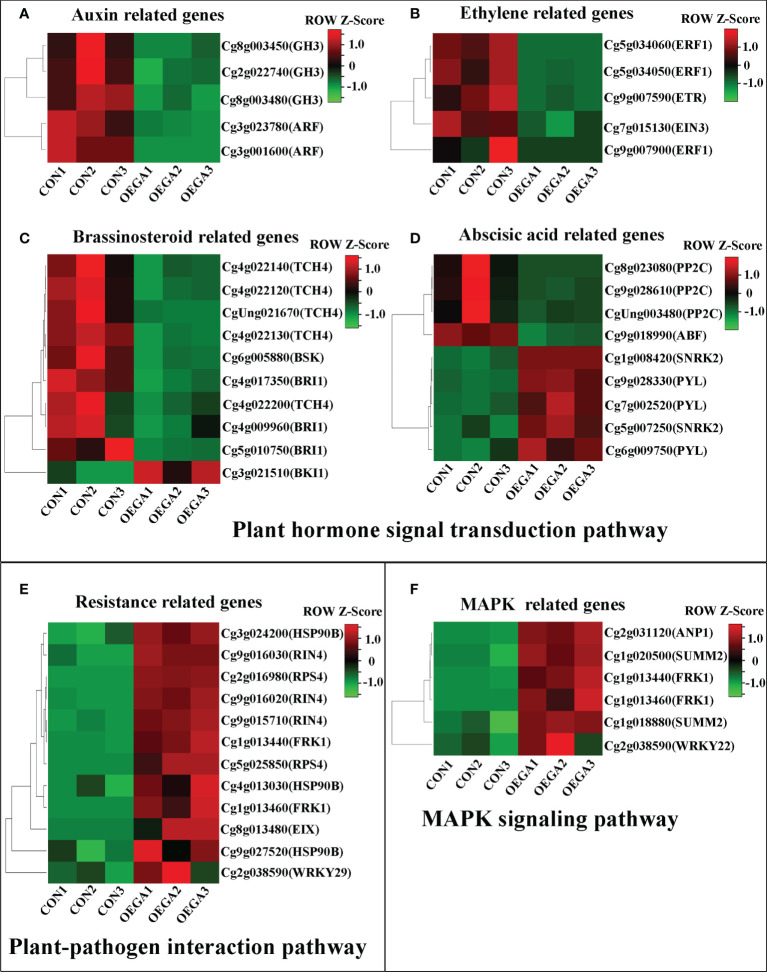
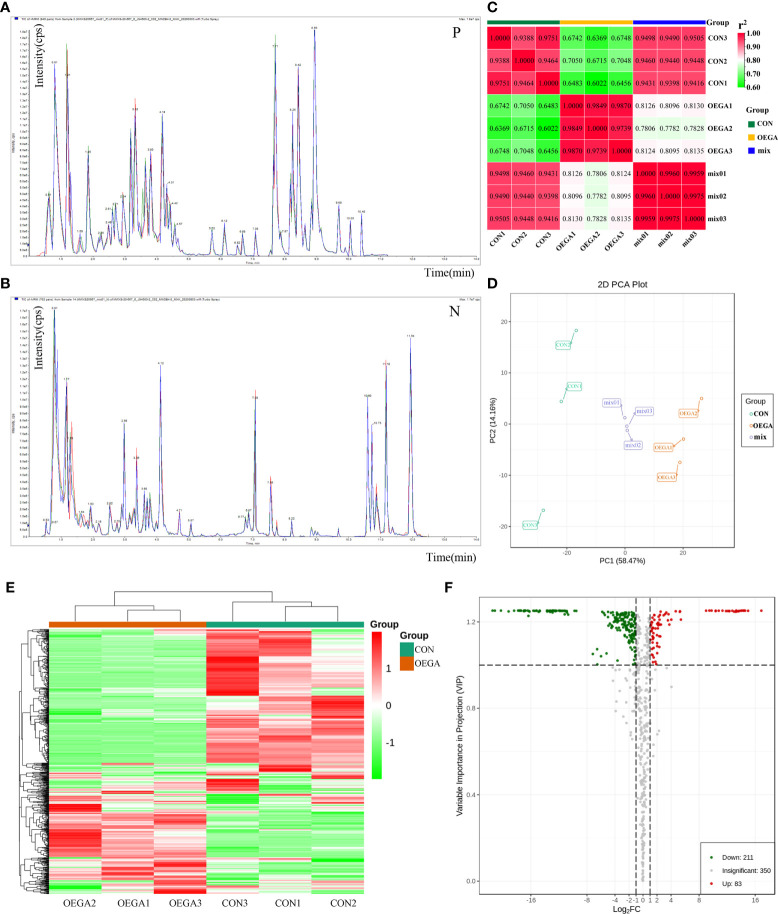
Similar articles
-
Transcriptomic and Metabolomic Insights into Plant Hormone Modulation and Secondary Metabolite Accumulation in Basil Under Far-Red and Ultraviolet-A Light.Int J Mol Sci. 2025 Apr 16;26(8):3758. doi: 10.3390/ijms26083758. Int J Mol Sci. 2025. PMID: 40332413 Free PMC article.
-
Integrative Analysis of the Transcriptome and Metabolome Reveals Genes Involved in Phenylpropanoid and Flavonoid Biosynthesis in the Trapa bispinosa Roxb.Front Plant Sci. 2022 Jul 7;13:913265. doi: 10.3389/fpls.2022.913265. eCollection 2022. Front Plant Sci. 2022. PMID: 35873984 Free PMC article.
-
Comparative transcriptome and metabolome analysis revealed diversity in the response of resistant and susceptible rose (Rosa hybrida) varieties to Marssonina rosae.Front Plant Sci. 2024 Feb 22;15:1362287. doi: 10.3389/fpls.2024.1362287. eCollection 2024. Front Plant Sci. 2024. PMID: 38455733 Free PMC article.
-
Multi-omics analysis reveals the molecular changes accompanying heavy-grazing-induced dwarfing of Stipa grandis.Front Plant Sci. 2022 Oct 4;13:995074. doi: 10.3389/fpls.2022.995074. eCollection 2022. Front Plant Sci. 2022. PMID: 36407579 Free PMC article.
-
Combined transcriptome and metabolome analysis of Nerium indicum L. elaborates the key pathways that are activated in response to witches' broom disease.BMC Plant Biol. 2022 Jun 14;22(1):291. doi: 10.1186/s12870-022-03672-z. BMC Plant Biol. 2022. PMID: 35701735 Free PMC article.
References
-
- Alonso-Ramírez A., Rodríguez D., Reyes D., Jiménez J. A., Nicolás G., López-Climent M., et al. . (2009). Evidence for a role of gibberellins in salicylic acid-modulated early plant responses to abiotic stress in arabidopsis seeds. Plant Physiol. 150 (3), 1335–1344. doi: 10.1104/pp.109.139352 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources