

ER stress as a trigger of UPR and ER-phagy in cancer growth and spread
- PMID: 36408145
- PMCID: PMC9667062
- DOI: 10.3389/fonc.2022.997235
ER stress as a trigger of UPR and ER-phagy in cancer growth and spread
Abstract
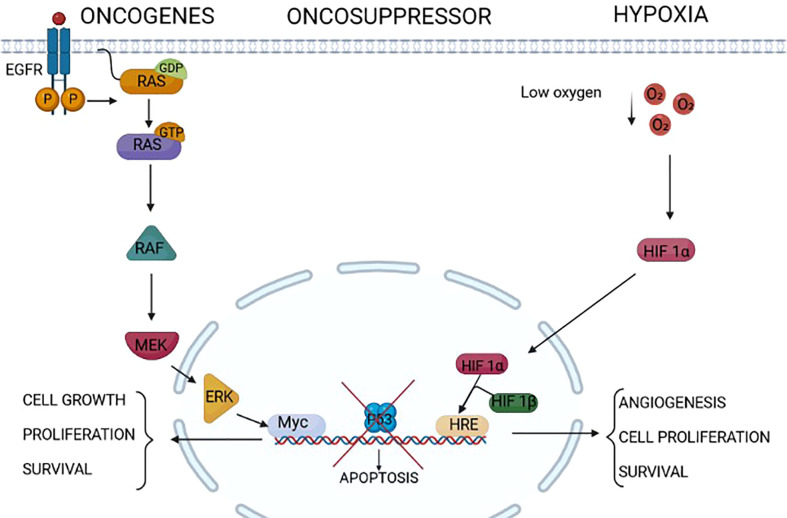
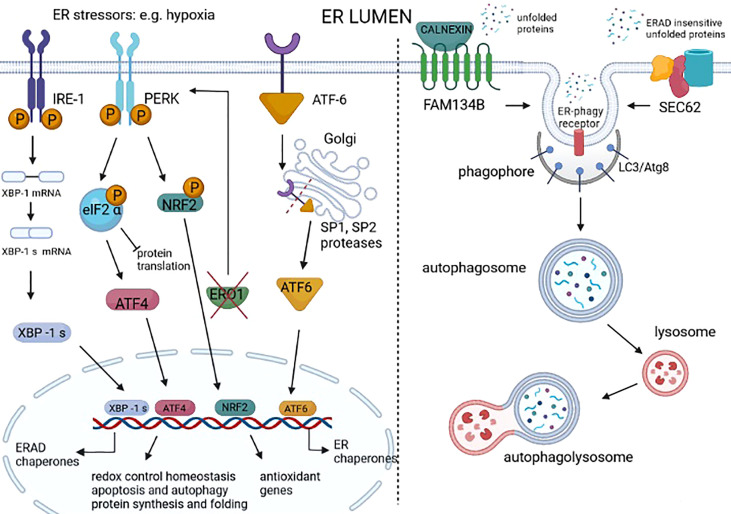
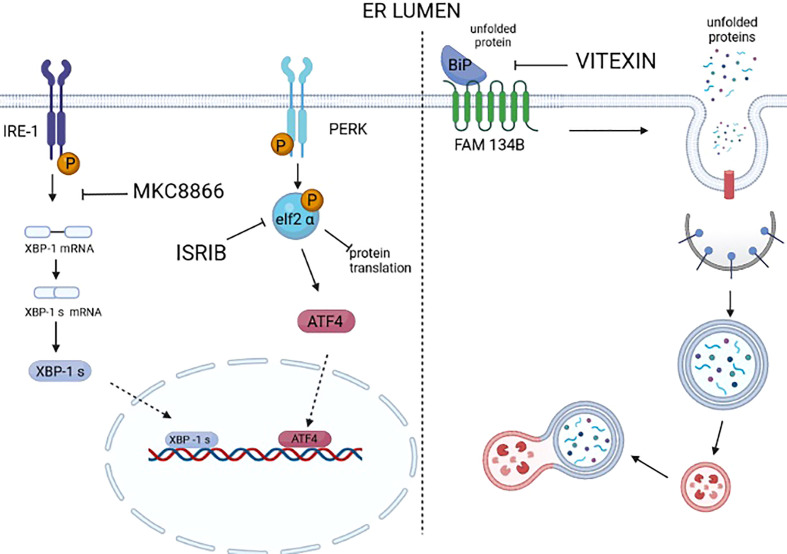
Tumors can survive environmental and metabolic stress by triggering homeostatic responses that re-establish the pre-stress status and permit them to grow and thrive. The endoplasmic reticulum (ER) is the organelle where proteins undergo post-translational modifications and are folded and exported to the secretory pathway. Its environment and activity are therefore fundamental for proteostasis, i.e., the plethora of mechanisms controlling protein formation, folding, degradation, and secretion, needed to assure protein balance and cellular health. In different tumor-related conditions, such as after the activation of oncogenes or under hypoxia and nutrient deprivation, the ER experiences stress, triggered by a high load of proteins to be folded compared to the limited folding capacity of the organelle. As a consequence, three ER membrane sensors and the related unfolded protein response (UPR) are activated. The UPR comprises a complex interconnection between signal transduction pathways that promote a homeostatic response that acts by increasing the amount of protein chaperones and of proteins involved in ER-associated protein degradation (ERAD) on one hand and attenuating protein translation on the other. ER-phagy, literally "eating" the ER, is part of another homeostatic response consisting of the clearance of non-functional ER portions including misfolded proteins. This response is also activated by a set of dedicated ER-phagy receptors after ER stimuli, which overlap the stimuli generating ER stress. Thus, the UPR and ER-phagy are two closely related homeostatic mechanisms that cooperate in re-establishing ER homeostasis. However, while the role of the UPR in favoring cancer growth and thriving by promoting angiogenesis, metastasis, chemotherapy resistance, and epithelial-to-mesenchymal transition is consolidated, that of ER-phagy is still in its infancy. This essay provides an overview of emerging concepts on ER stress, the UPR, and ER-phagy and their crosstalk in tumorigenesis. We also critically review new findings on their pharmacological targeting in cancer.
Keywords: ER stress; ER-phagy; ERO1 alpha; UPR; cancer; hypoxia.
Copyright © 2022 Cherubini and Zito.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD.Dev Cell. 2021 Apr 5;56(7):949-966. doi: 10.1016/j.devcel.2021.03.005. Epub 2021 Mar 24. Dev Cell. 2021. PMID: 33765438 Review.
-
Proteostasis In The Endoplasmic Reticulum: Road to Cure.Cancers (Basel). 2019 Nov 14;11(11):1793. doi: 10.3390/cancers11111793. Cancers (Basel). 2019. PMID: 31739582 Free PMC article. Review.
-
Endoplasmic Reticulum (ER) and ER-Phagy.Prog Mol Subcell Biol. 2021;59:99-114. doi: 10.1007/978-3-030-67696-4_5. Prog Mol Subcell Biol. 2021. PMID: 34050863
-
How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases.Int J Mol Sci. 2021 Feb 19;22(4):2078. doi: 10.3390/ijms22042078. Int J Mol Sci. 2021. PMID: 33669844 Free PMC article. Review.
-
Mechanisms of Endoplasmic Reticulum Protein Homeostasis in Plants.Int J Mol Sci. 2023 Dec 18;24(24):17599. doi: 10.3390/ijms242417599. Int J Mol Sci. 2023. PMID: 38139432 Free PMC article. Review.
Cited by
-
Modulation of Endoplasmic Reticulum Stress in Experimental Anti-Cancer Therapy.Int J Mol Sci. 2025 Jul 3;26(13):6407. doi: 10.3390/ijms26136407. Int J Mol Sci. 2025. PMID: 40650182 Free PMC article. Review.
-
LncRNAs and CircRNAs in Endoplasmic Reticulum Stress: A Promising Target for Cardiovascular Disease?Int J Mol Sci. 2023 Jun 8;24(12):9888. doi: 10.3390/ijms24129888. Int J Mol Sci. 2023. PMID: 37373035 Free PMC article. Review.
-
Investigation of the mutual crosstalk between ER stress and PI3K/AKT/mTOR signaling pathway in iron overload-induced liver injury in chicks.Biometals. 2024 Aug;37(4):955-969. doi: 10.1007/s10534-024-00588-z. Epub 2024 Mar 14. Biometals. 2024. PMID: 38483766
-
Bibliometric analysis of autophagy in NAFLD from 2004 to 2023.Medicine (Baltimore). 2024 Dec 6;103(49):e40835. doi: 10.1097/MD.0000000000040835. Medicine (Baltimore). 2024. PMID: 39654183 Free PMC article.
-
Organellar quality control crosstalk in aging-related disease: Innovation to pave the way.Aging Cell. 2025 Jan;24(1):e14447. doi: 10.1111/acel.14447. Epub 2024 Dec 12. Aging Cell. 2025. PMID: 39668579 Free PMC article. Review.
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials