

Ribosomal RNA operons define a central functional compartment in the Streptomyces chromosome
- PMID: 36408918
- PMCID: PMC9723626
- DOI: 10.1093/nar/gkac1076
Ribosomal RNA operons define a central functional compartment in the Streptomyces chromosome
Abstract
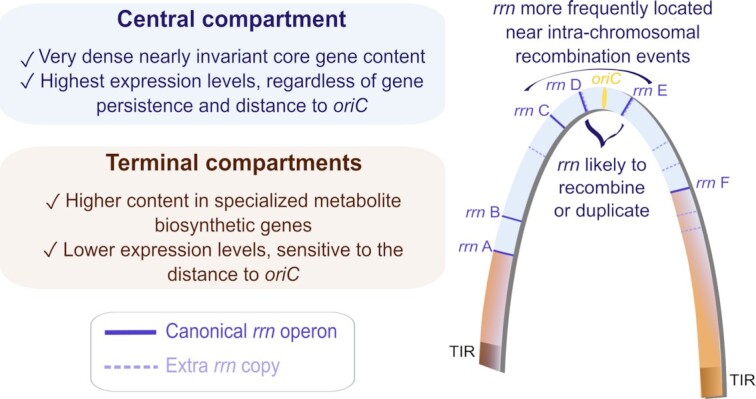
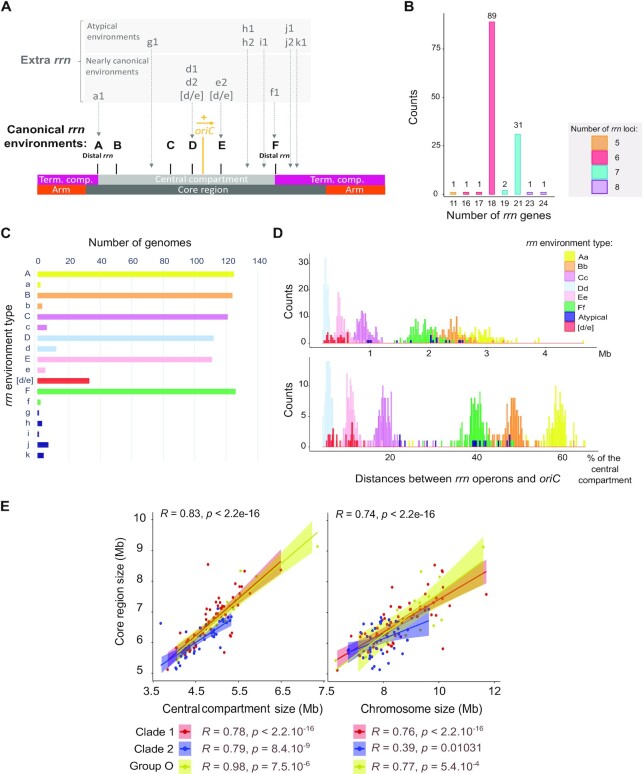
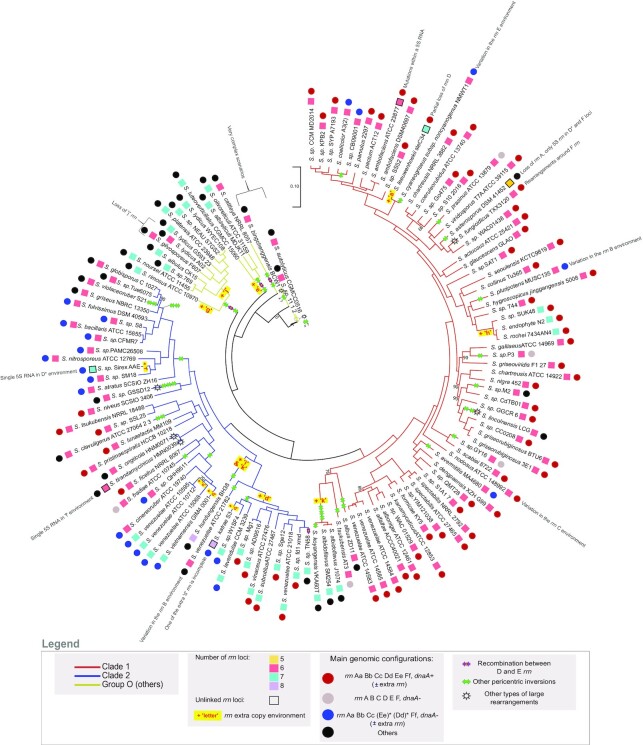
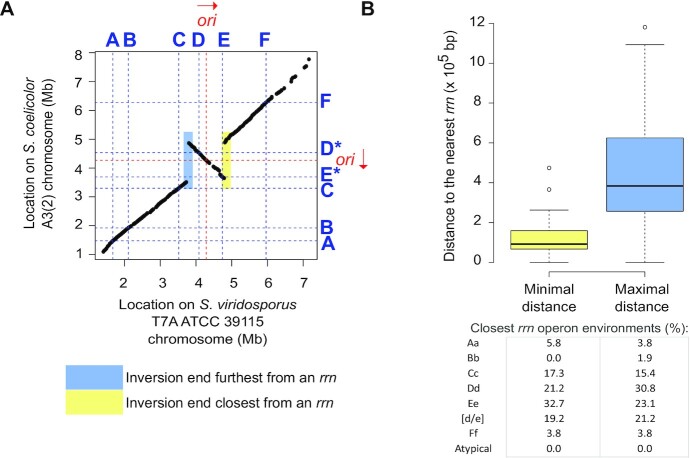
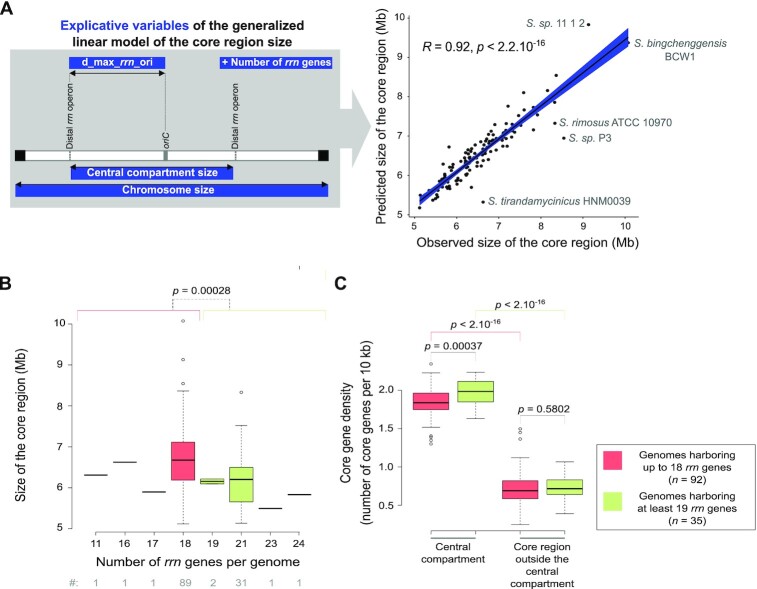
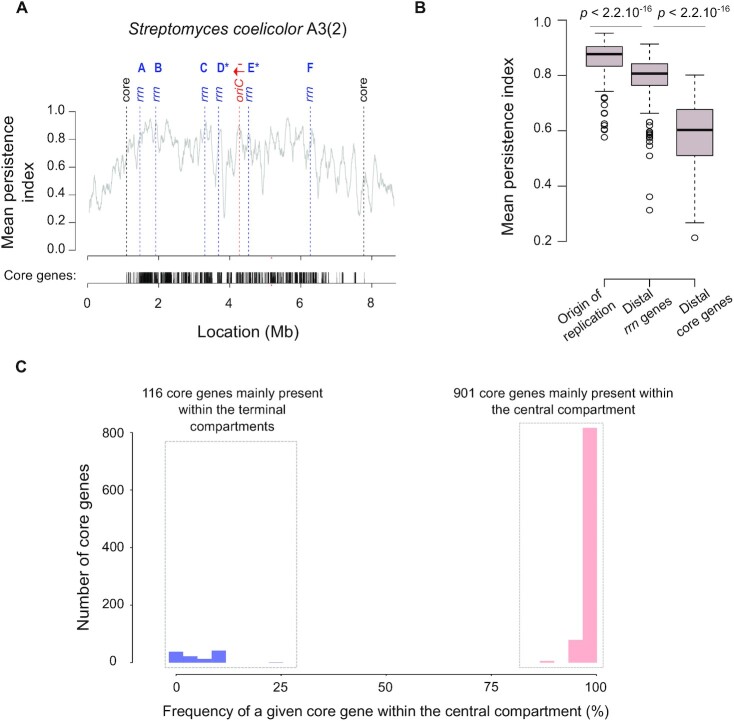
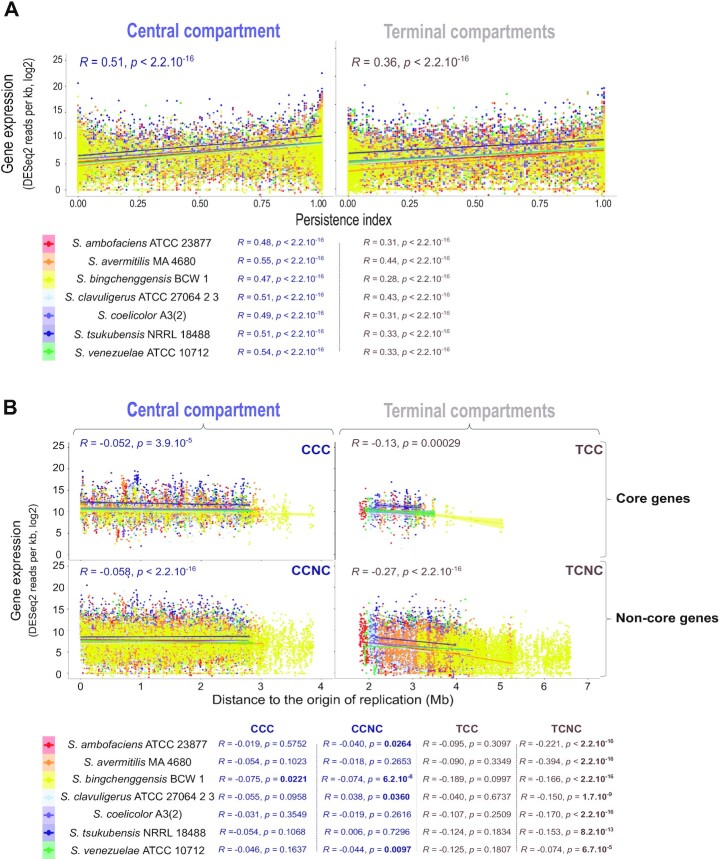
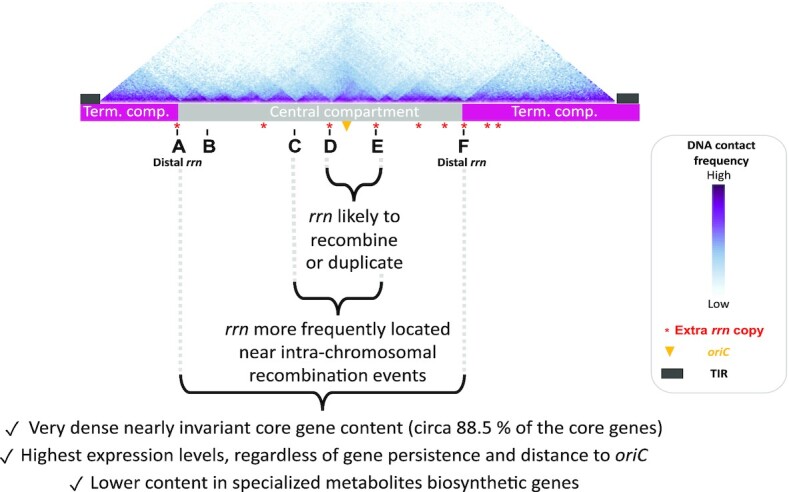
Streptomyces are prolific producers of specialized metabolites with applications in medicine and agriculture. These bacteria possess a large linear chromosome genetically compartmentalized: core genes are grouped in the central part, while terminal regions are populated by poorly conserved genes. In exponentially growing cells, chromosome conformation capture unveiled sharp boundaries formed by ribosomal RNA (rrn) operons that segment the chromosome into multiple domains. Here we further explore the link between the genetic distribution of rrn operons and Streptomyces genetic compartmentalization. A large panel of genomes of species representative of the genus diversity revealed that rrn operons and core genes form a central skeleton, the former being identifiable from their core gene environment. We implemented a new nomenclature for Streptomyces genomes and trace their rrn-based evolutionary history. Remarkably, rrn operons are close to pericentric inversions. Moreover, the central compartment delimited by rrn operons has a very dense, nearly invariant core gene content. Finally, this compartment harbors genes with the highest expression levels, regardless of gene persistence and distance to the origin of replication. Our results highlight that rrn operons are structural boundaries of a central functional compartment prone to transcription in Streptomyces.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Homologous recombination between rrn operons rearranges the chromosome in host-specialized species of Salmonella.FEMS Microbiol Lett. 1998 Jul 15;164(2):275-81. doi: 10.1111/j.1574-6968.1998.tb13098.x. FEMS Microbiol Lett. 1998. PMID: 9682477
-
Bacteria can maintain rRNA operons solely on plasmids for hundreds of millions of years.Nat Commun. 2023 Nov 14;14(1):7232. doi: 10.1038/s41467-023-42681-w. Nat Commun. 2023. PMID: 37963895 Free PMC article.
-
Dynamics of the compartmentalized Streptomyces chromosome during metabolic differentiation.Nat Commun. 2021 Sep 1;12(1):5221. doi: 10.1038/s41467-021-25462-1. Nat Commun. 2021. PMID: 34471117 Free PMC article.
-
Dynamics of the Streptomyces chromosome: chance and necessity.Trends Genet. 2023 Nov;39(11):873-887. doi: 10.1016/j.tig.2023.07.008. Epub 2023 Sep 6. Trends Genet. 2023. PMID: 37679290 Review.
-
Chromosomal rearrangements in enteric bacteria.Electrophoresis. 1998 Apr;19(4):569-72. doi: 10.1002/elps.1150190417. Electrophoresis. 1998. PMID: 9588803 Review.
Cited by
-
A treasure trove of 1034 actinomycete genomes.Nucleic Acids Res. 2024 Jul 22;52(13):7487-7503. doi: 10.1093/nar/gkae523. Nucleic Acids Res. 2024. PMID: 38908028 Free PMC article.
-
The distinct cell physiology of Bradyrhizobium at the population and cellular level.BMC Microbiol. 2024 Apr 20;24(1):129. doi: 10.1186/s12866-024-03272-x. BMC Microbiol. 2024. PMID: 38643099 Free PMC article.
-
Title-Evaluation of Interplay of Gene Expression and Chromosome Structure in E. coli Growth: Regulatory Insights.Curr Microbiol. 2024 Jun 21;81(8):235. doi: 10.1007/s00284-024-03773-y. Curr Microbiol. 2024. PMID: 38907057 No abstract available.
-
Prophage induction can facilitate the in vitro dispersal of multicellular Streptomyces structures.PLoS Biol. 2024 Jul 25;22(7):e3002725. doi: 10.1371/journal.pbio.3002725. eCollection 2024 Jul. PLoS Biol. 2024. PMID: 39052683 Free PMC article.
References
-
- Berdy J. Thoughts and facts about antibiotics: where we are now and where we are heading. J. Antibiot. (Tokyo). 2012; 65:441. - PubMed
-
- Redenbach M., Kieser H.M., Denapaite D., Eichner A., Cullum J., Kinashi H., Hopwood D.A.. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomycescoelicolor A3(2) chromosome. Mol. Microbiol. 1996; 21:77–96. - PubMed
-
- Omura S., Ikeda H., Ishikawa J., Hanamoto A., Takahashi C., Shinose M., Takahashi Y., Horikawa H., Nakazawa H., Osonoe T.et al. .. Genome sequence of an industrial microorganism Streptomycesavermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:12215–12220. - PMC - PubMed
-
- Karoonuthaisiri N., Weaver D., Huang J., Cohen S.N., Kao C.M.. Regional organization of gene expression in Streptomycescoelicolor. Gene. 2005; 353:53–66. - PubMed
-
- Ikeda H., Ishikawa J., Hanamoto A., Shinose M., Kikuchi H., Shiba T., Sakaki Y., Hattori M., Omura S.. Complete genome sequence and comparative analysis of the industrial microorganism Streptomycesavermitilis. Nat. Biotechnol. 2003; 21:526–531. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
