

Clinical and molecular characterization of a large primary hyperoxaluria cohort from Saudi Arabia: a retrospective study
- PMID: 36409364
- PMCID: PMC10154271
- DOI: 10.1007/s00467-022-05784-y
Clinical and molecular characterization of a large primary hyperoxaluria cohort from Saudi Arabia: a retrospective study
Abstract
Background: Primary hyperoxalurias (PHs) constitute rare disorders resulting in abnormal glyoxalate metabolism. PH-associated phenotypes range from progressive nephrocalcinosis and/or recurrent urolithiasis to early kidney failure.
Methods: A retrospective study was conducted for patients with confirmed PH diagnoses from three tertiary centers in Saudi Arabia. Detailed clinical molecular diagnosis was performed for 25 affected individuals. Whole exome sequencing (WES)-based molecular diagnosis was performed for all affected individuals.
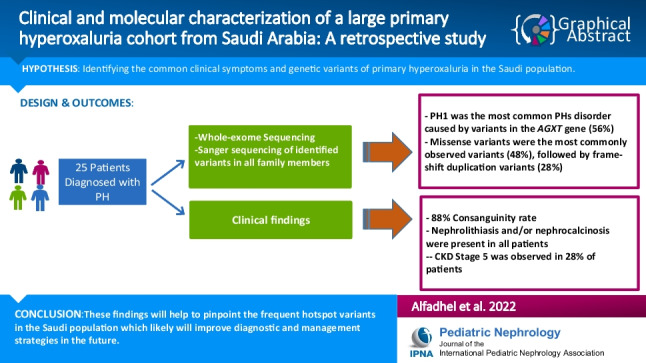
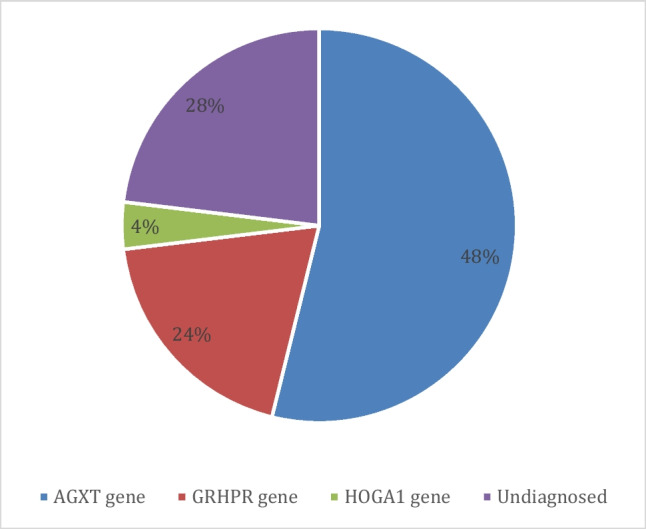
Results: The male:female ratio was 52% male (n = 13) and 48% female (n = 12), and consanguinity was present in 88%. Nephrolithiasis and/or nephrocalcinosis were present in all patients. Kidney stones were present in 72%, nephrocalcinosis in 60%, hematuria in 32%, proteinuria in 16%, abdominal pain in 36%, developmental delay in 8%, and chronic kidney disease stage 5 (CKD stage 5) was observed in 28% of the patients. The most common PH disorder was type I caused by variants in the AGXT gene, accounting for 56%. The GRHPR gene variants were identified in 4 patients, 16% of the total cases. Seven patients did not reveal any associated variants. Missense variants were the most commonly observed variants (48%), followed by frame-shift duplication variants (28%).
Conclusions: Characterization of the genetic and clinical aspects of PH in this unique population provides direction for improved patient management and further research. A higher resolution version of the Graphical abstract is available as Supplementary information.
Keywords: AGXT; GRHPR; Nephrocalcinosis; Primary hyperoxaluria; Saudi Arabia.
© 2022. The Author(s).
Conflict of interest statement
None.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
