

JAK-STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States
- PMID: 36409824
- PMCID: PMC9845989
- DOI: 10.1158/0008-5472.CAN-22-0423
JAK-STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States
Abstract
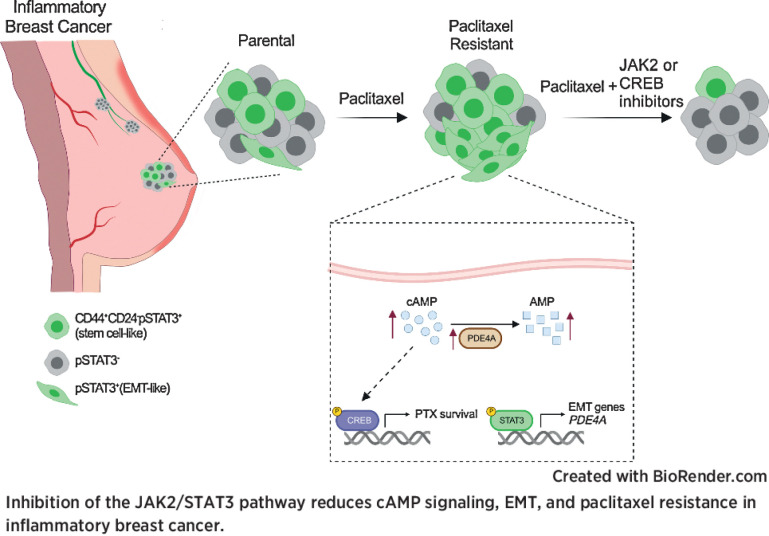
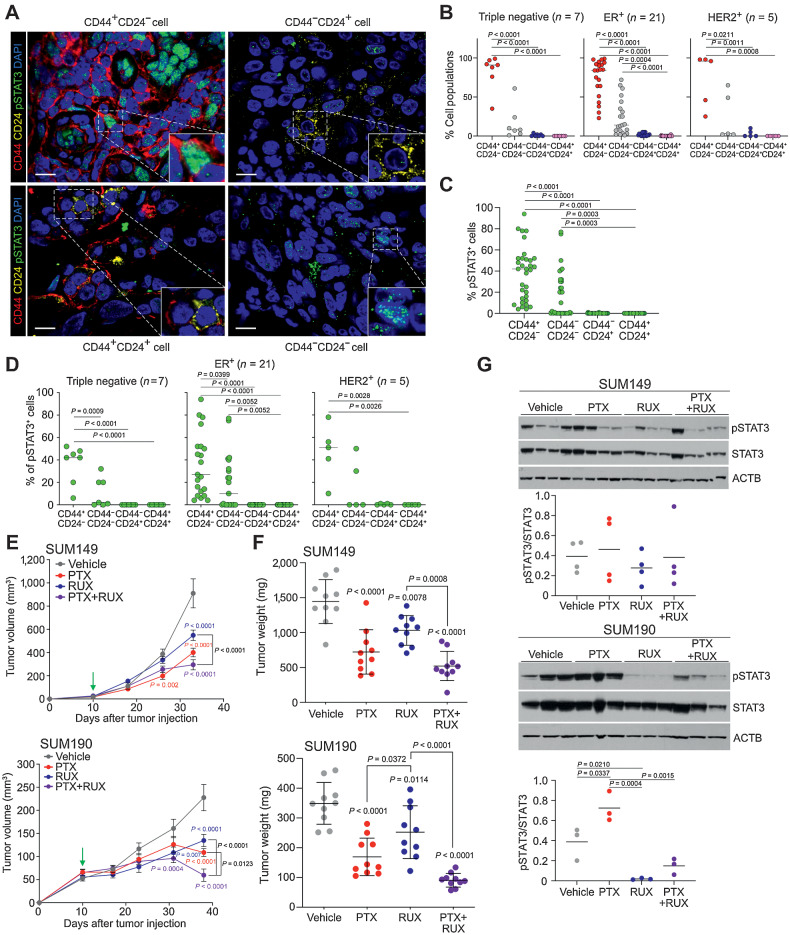
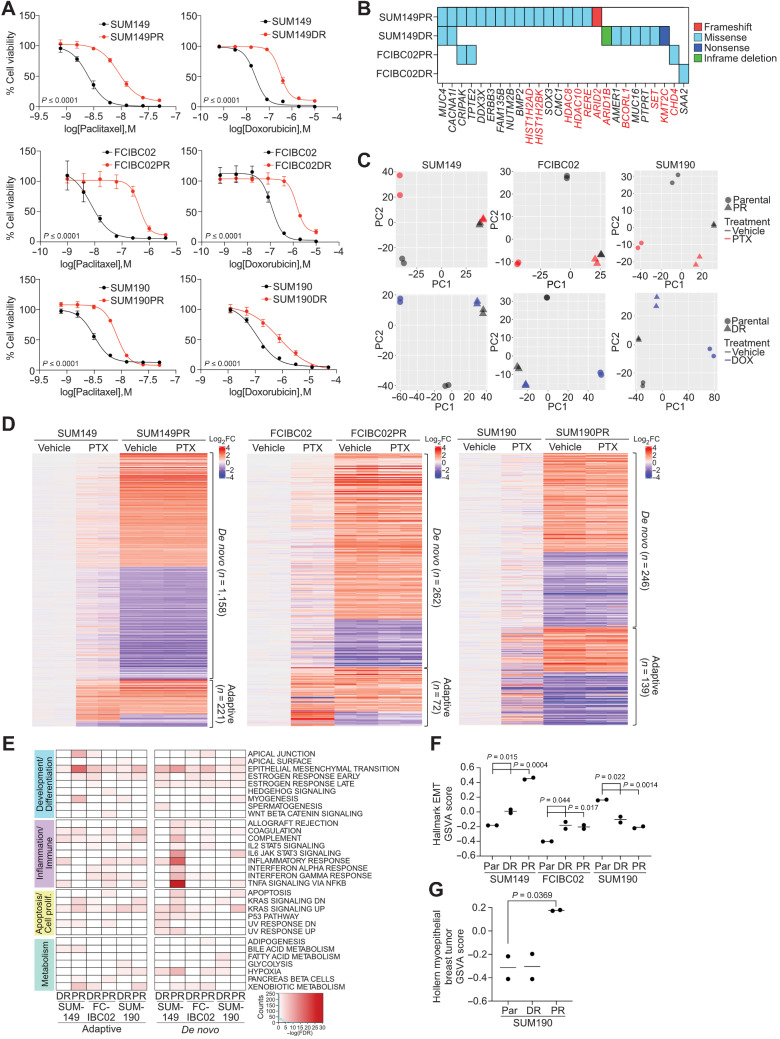
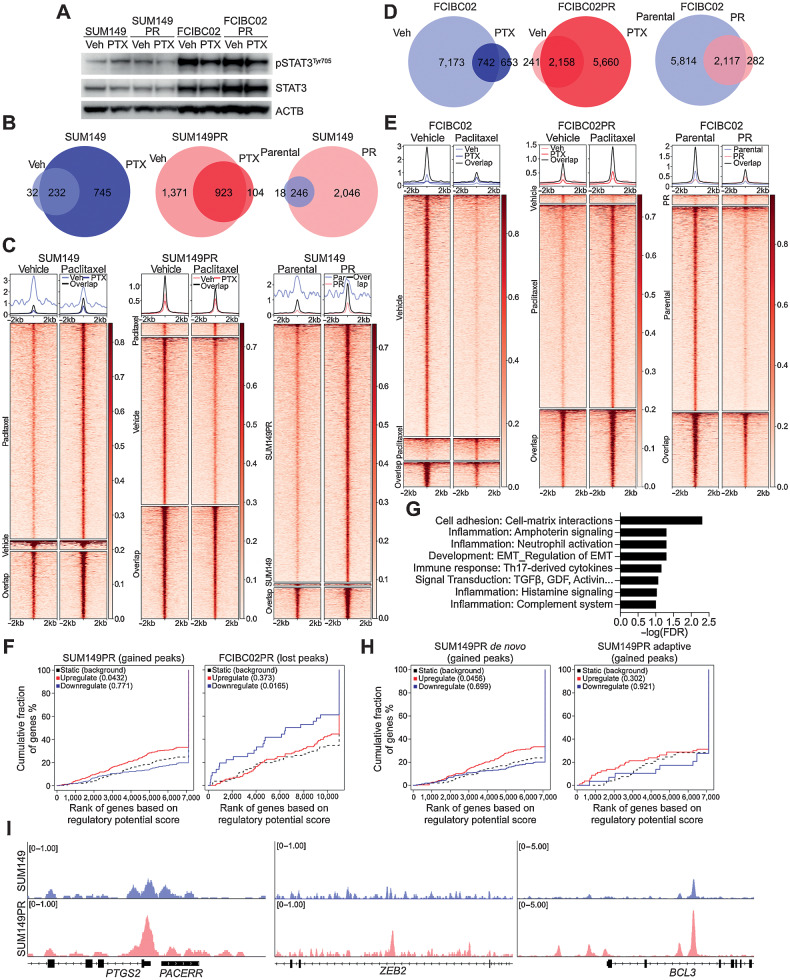
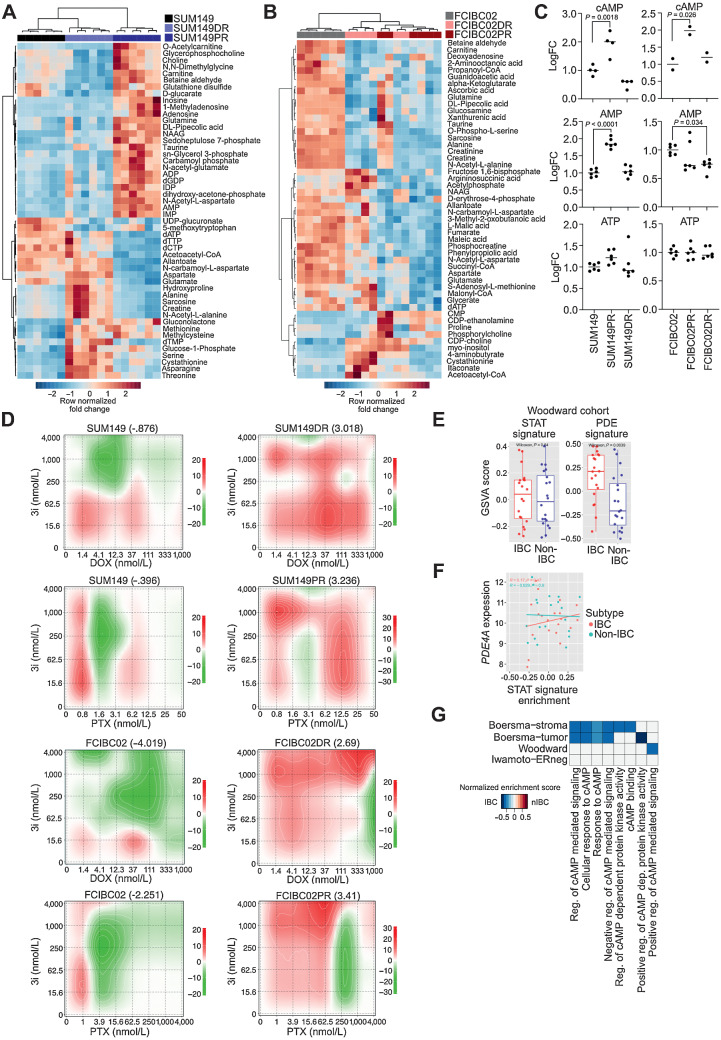
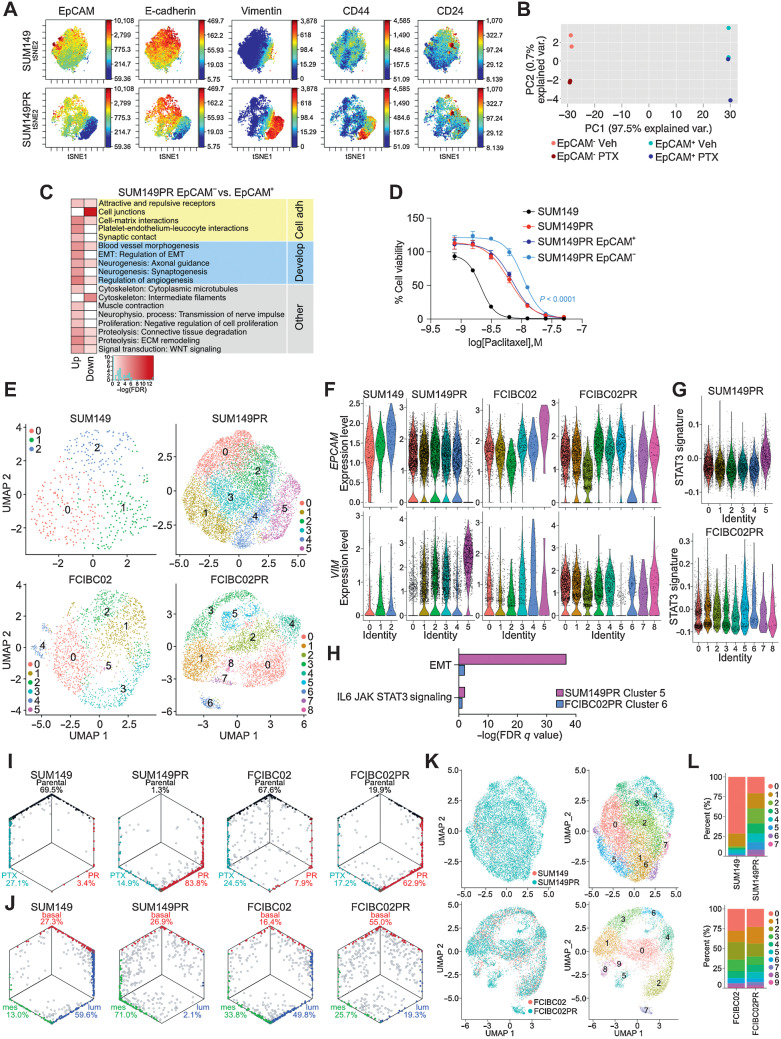
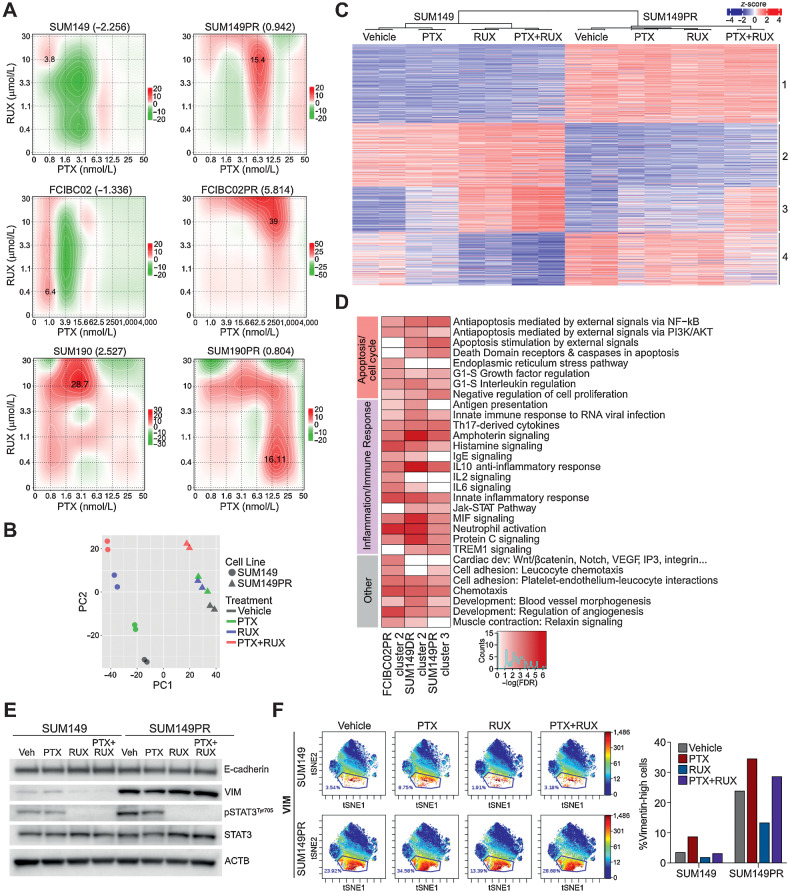
Inflammatory breast cancer (IBC) is a difficult-to-treat disease with poor clinical outcomes due to high risk of metastasis and resistance to treatment. In breast cancer, CD44+CD24- cells possess stem cell-like features and contribute to disease progression, and we previously described a CD44+CD24-pSTAT3+ breast cancer cell subpopulation that is dependent on JAK2/STAT3 signaling. Here we report that CD44+CD24- cells are the most frequent cell type in IBC and are commonly pSTAT3+. Combination of JAK2/STAT3 inhibition with paclitaxel decreased IBC xenograft growth more than either agent alone. IBC cell lines resistant to paclitaxel and doxorubicin were developed and characterized to mimic therapeutic resistance in patients. Multi-omic profiling of parental and resistant cells revealed enrichment of genes associated with lineage identity and inflammation in chemotherapy-resistant derivatives. Integrated pSTAT3 chromatin immunoprecipitation sequencing and RNA sequencing (RNA-seq) analyses showed pSTAT3 regulates genes related to inflammation and epithelial-to-mesenchymal transition (EMT) in resistant cells, as well as PDE4A, a cAMP-specific phosphodiesterase. Metabolomic characterization identified elevated cAMP signaling and CREB as a candidate therapeutic target in IBC. Investigation of cellular dynamics and heterogeneity at the single cell level during chemotherapy and acquired resistance by CyTOF and single cell RNA-seq identified mechanisms of resistance including a shift from luminal to basal/mesenchymal cell states through selection for rare preexisting subpopulations or an acquired change. Finally, combination treatment with paclitaxel and JAK2/STAT3 inhibition prevented the emergence of the mesenchymal chemo-resistant subpopulation. These results provide mechanistic rational for combination of chemotherapy with inhibition of JAK2/STAT3 signaling as a more effective therapeutic strategy in IBC.
Significance: Chemotherapy resistance in inflammatory breast cancer is driven by the JAK2/STAT3 pathway, in part via cAMP/PKA signaling and a cell state switch, which can be overcome using paclitaxel combined with JAK2 inhibitors.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Abraham HG, Xia Y, Mukherjee B, Merajver SD. Incidence and survival of inflammatory breast cancer between 1973 and 2015 in the SEER database. Breast Cancer Res Treat 2021;185:229–38. - PubMed
-
- Bertucci F, Finetti P, Rougemont J, Charafe-Jauffret E, Nasser V, Loriod B, et al. Gene expression profiling for molecular characterization of inflammatory breast cancer and prediction of response to chemotherapy. Cancer Res 2004;64:8558–65. - PubMed
-
- Vermeulen PB, van Golen KL, Dirix LY. Angiogenesis, lymphangiogenesis, growth pattern, and tumor emboli in inflammatory breast cancer: a review of the current knowledge. Cancer 2010;116:2748–54. - PubMed
-
- Kleer CG, Griffith KA, Sabel MS, Gallagher G, van Golen KL, Wu ZF, et al. RhoC-GTPase is a novel tissue biomarker associated with biologically aggressive carcinomas of the breast. Breast Cancer Res Treat 2005;93:101–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
