

How vascular smooth muscle cell phenotype switching contributes to vascular disease
- PMID: 36411459
- PMCID: PMC9677683
- DOI: 10.1186/s12964-022-00993-2
How vascular smooth muscle cell phenotype switching contributes to vascular disease
Abstract
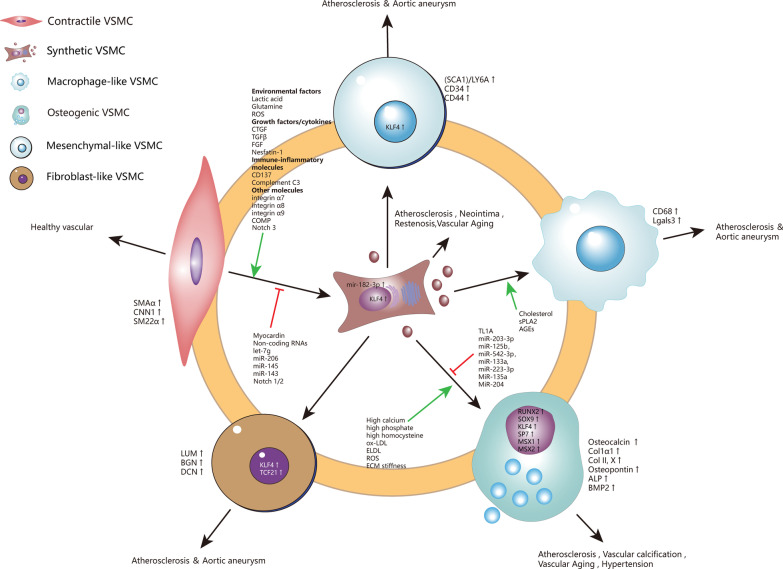
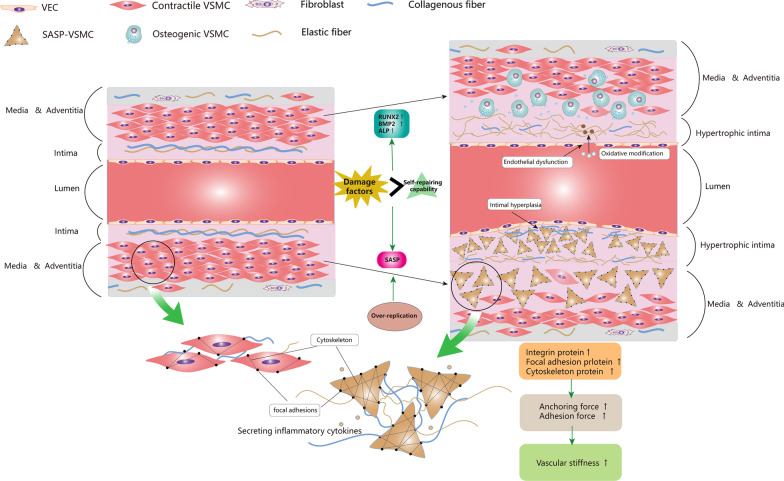
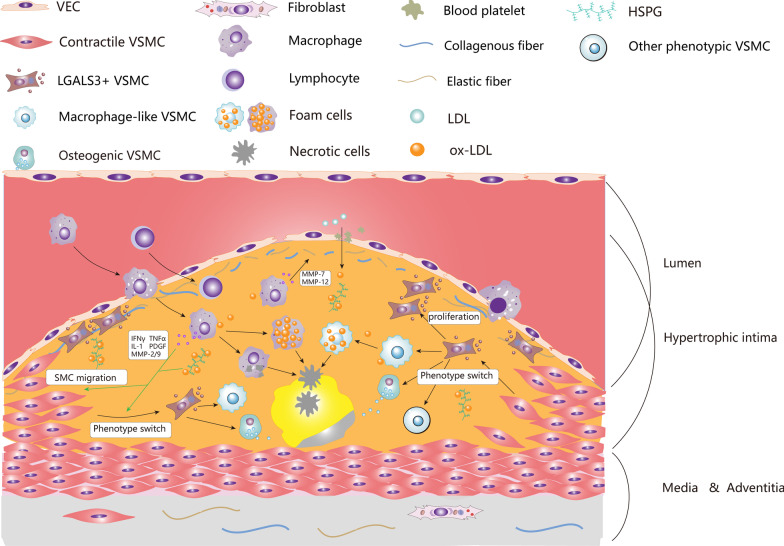
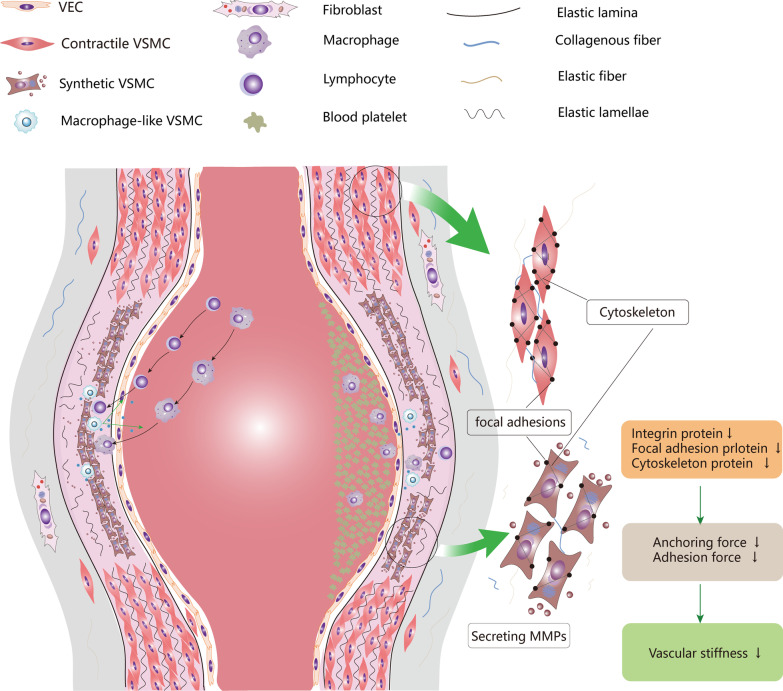
Vascular smooth muscle cells (VSMCs) are the most abundant cell in vessels. Earlier experiments have found that VSMCs possess high plasticity. Vascular injury stimulates VSMCs to switch into a dedifferentiated type, also known as synthetic VSMCs, with a high migration and proliferation capacity for repairing vascular injury. In recent years, largely owing to rapid technological advances in single-cell sequencing and cell-lineage tracing techniques, multiple VSMCs phenotypes have been uncovered in vascular aging, atherosclerosis (AS), aortic aneurysm (AA), etc. These VSMCs all down-regulate contractile proteins such as α-SMA and calponin1, and obtain specific markers and similar cellular functions of osteoblast, fibroblast, macrophage, and mesenchymal cells. This highly plastic phenotype transformation is regulated by a complex network consisting of circulating plasma substances, transcription factors, growth factors, inflammatory factors, non-coding RNAs, integrin family, and Notch pathway. This review focuses on phenotypic characteristics, molecular profile and the functional role of VSMCs phenotype landscape; the molecular mechanism regulating VSMCs phenotype switching; and the contribution of VSMCs phenotype switching to vascular aging, AS, and AA. Video Abstract.
Keywords: Aortic aneurysm; Atherosclerosis; Phenotype switching; Vascular aging; Vascular smooth muscle cells.
© 2022. The Author(s).
Conflict of interest statement
I declare that there is no competing interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
