

Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria
- PMID: 36413997
- PMCID: PMC9748256
- DOI: 10.1016/j.ajhg.2022.10.013
Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria
Abstract
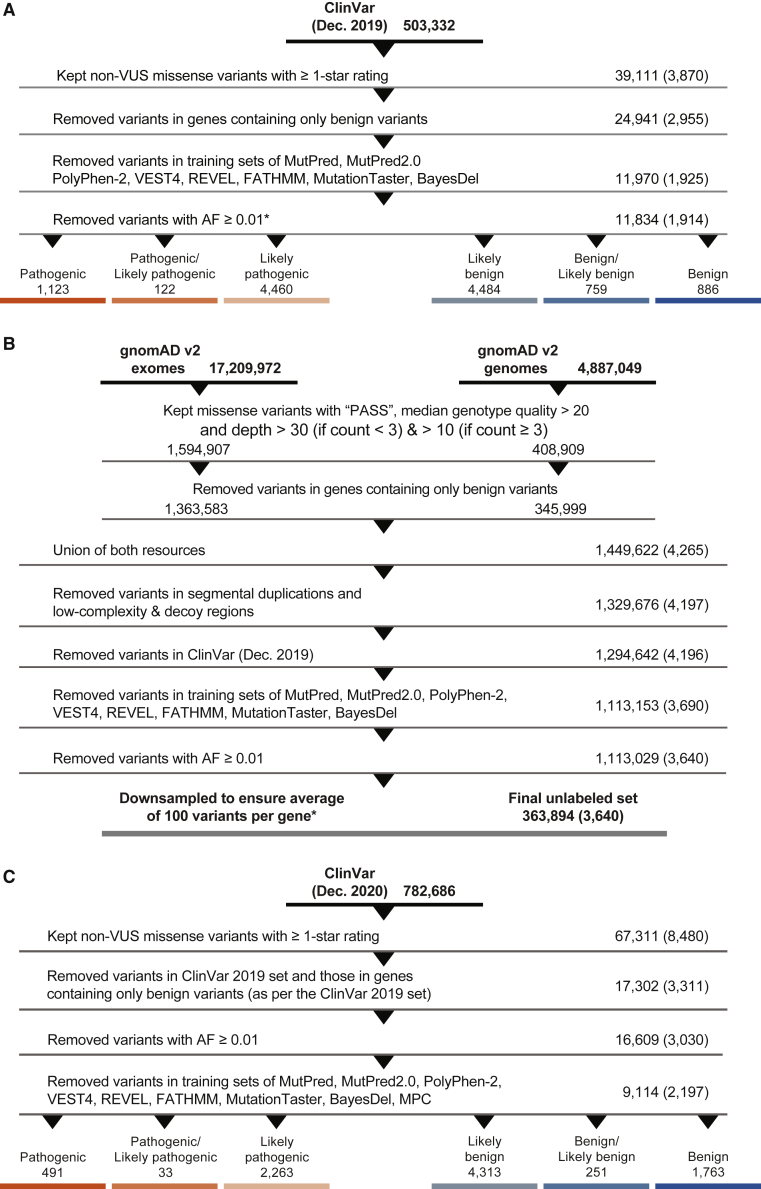
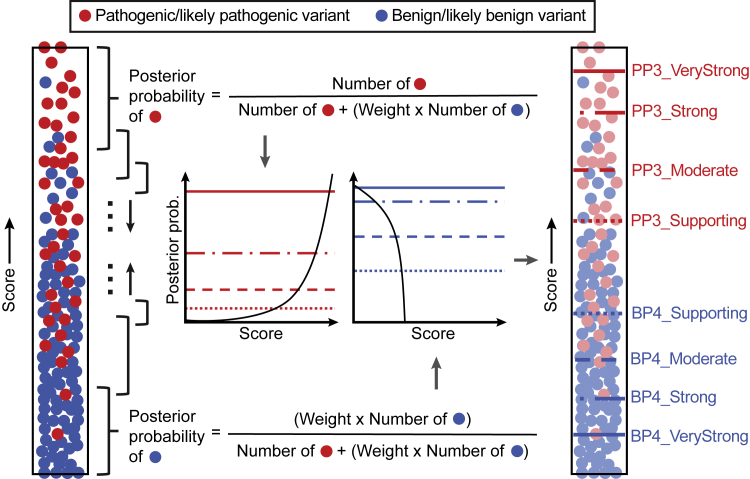
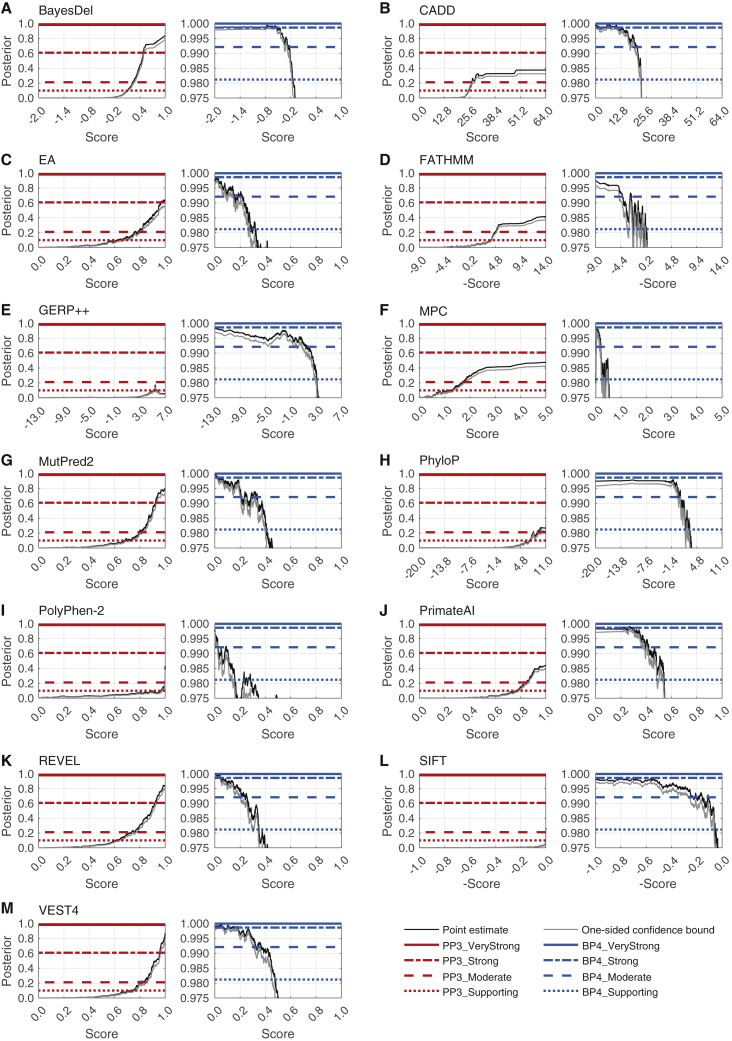
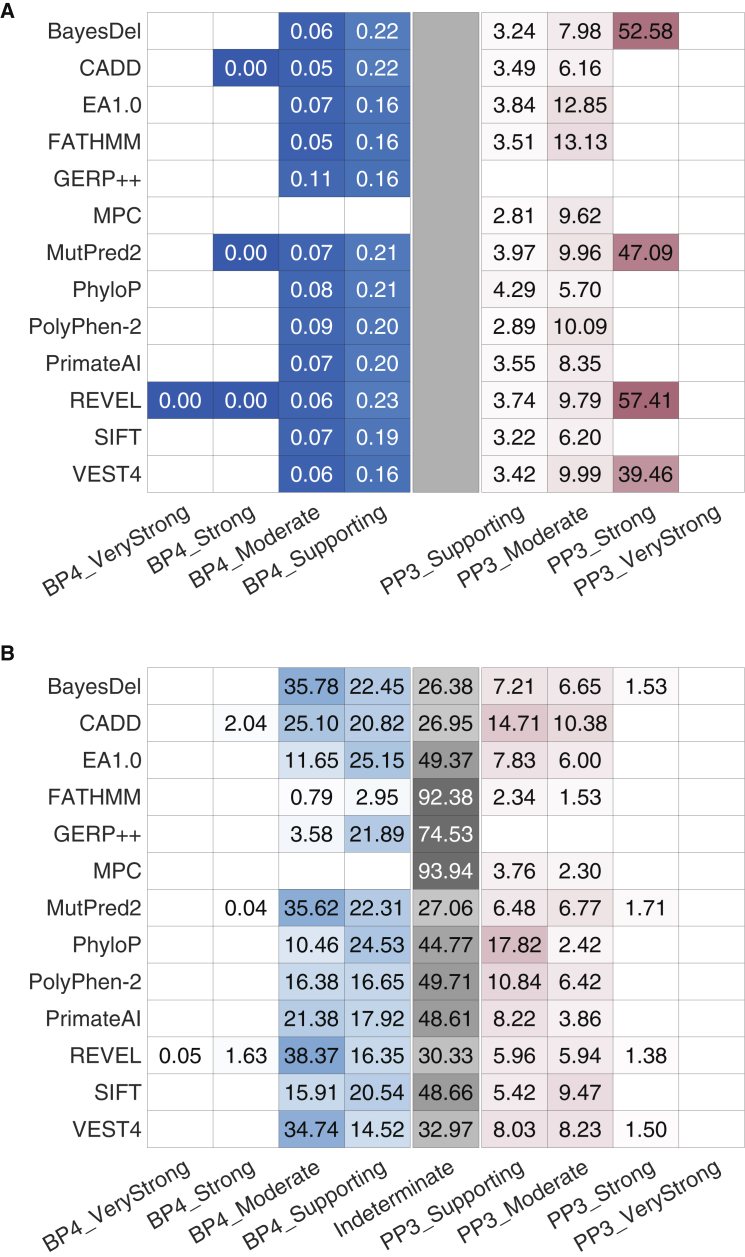
Recommendations from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) for interpreting sequence variants specify the use of computational predictors as "supporting" level of evidence for pathogenicity or benignity using criteria PP3 and BP4, respectively. However, score intervals defined by tool developers, and ACMG/AMP recommendations that require the consensus of multiple predictors, lack quantitative support. Previously, we described a probabilistic framework that quantified the strengths of evidence (supporting, moderate, strong, very strong) within ACMG/AMP recommendations. We have extended this framework to computational predictors and introduce a new standard that converts a tool's scores to PP3 and BP4 evidence strengths. Our approach is based on estimating the local positive predictive value and can calibrate any computational tool or other continuous-scale evidence on any variant type. We estimate thresholds (score intervals) corresponding to each strength of evidence for pathogenicity and benignity for thirteen missense variant interpretation tools, using carefully assembled independent data sets. Most tools achieved supporting evidence level for both pathogenic and benign classification using newly established thresholds. Multiple tools reached score thresholds justifying moderate and several reached strong evidence levels. One tool reached very strong evidence level for benign classification on some variants. Based on these findings, we provide recommendations for evidence-based revisions of the PP3 and BP4 ACMG/AMP criteria using individual tools and future assessment of computational methods for clinical interpretation.
Keywords: ACMG/AMP recommendations; PP3/BP4 criteria; clinical classification; computational predictors; in silico tools; likelihood ratio; posterior probability; variant interpretation.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The PERCH software, for which B.-J.F. is the inventor, has been non-exclusively licensed to Ambry Genetics Corporation for their clinical genetic testing services and research. B.-J.F. also reports funding and sponsorship to his institution on his behalf from Pfizer Inc., Regeneron Genetics Center LLC., and Astra Zeneca. A.O’D.-L. is a compensated member of the Scientific Advisory Board of Congenica. L.G.B. is an uncompensated member of the Illumina Medical Ethics committee and receives honoraria from Cold Spring Harbor Laboratory Press. V.P., B.-J.F., K.A.P., S.D.M., R.K., A.O’D.-L., and P.R. participated in the development of some of the tools assessed in this study. While every care was taken to mitigate any potential biases in this work, these authors’ participation in method development is noted.
Figures
References
-
- McInnes G., Sharo A.G., Koleske M.L., Brown J.E.H., Norstad M., Adhikari A.N., Wang S., Brenner S.E., Halpern J., Koenig B.A., et al. Opportunities and challenges for the computational interpretation of rare variation in clinically important genes. Am. J. Hum. Genet. 2021;108:535–548. doi: 10.1016/j.ajhg.2021.03.003. - DOI - PMC - PubMed
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- UM1 HG008900/HG/NHGRI NIH HHS/United States
- U24 HG009649/HG/NHGRI NIH HHS/United States
- U01 HG012022/HG/NHGRI NIH HHS/United States
- U24 HG006834/HG/NHGRI NIH HHS/United States
- U24 HG007346/HG/NHGRI NIH HHS/United States
- R01 CA121245/CA/NCI NIH HHS/United States
- U24 HG011450/HG/NHGRI NIH HHS/United States
- ZIA HG200359/ImNIH/Intramural NIH HHS/United States
- U24 HG009650/HG/NHGRI NIH HHS/United States
- R01 CA264971/CA/NCI NIH HHS/United States
- K99 LM012992/LM/NLM NIH HHS/United States
- U24 CA258119/CA/NCI NIH HHS/United States
- U01 HG011755/HG/NHGRI NIH HHS/United States
- U41 HG009649/HG/NHGRI NIH HHS/United States
- R13 HG006650/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
